| [ начало ] | [ Г ] |
Глицерин
(хим., Glyc é rine фр., glycerin нем. и англ.) С 2 Н 3 О 2 = С 2 Н 5 (ОН) 2 — открыт в 1779 г. Шееле, заметившим, что при кипячении оливкового масла с глеnом, кроме свинцового пластыря (свинцового мыла, т. е. свинцовой соли жирных кислот), получается еще сладкая, сиропообразная жидкость; тем же способом Шееле получил затем Г. из масел: миндального, льняного, сурепного, коровьего и из свиного жира. Почти верный процентный (элементарный) состав Г. дан Шеврелем (1813 г.), который доказал, что, как упомянутые выше растительные масла, так и животные жиры (см.) по химическому характеру можно считать кислотными эфирами Г., и таким образом верно определил алкогольную натуру Г. (см. Алкоголи). Окончательно этот взгляд утвержден опытами Бертело (1853 и 185 4 гг.), получившим искусственно жиры нагреванием Г. с жирными кислотами; образование эфиров с одним, двумя и тремя эквивалентами взятой кислоты (смотря по условиям опыта) установило трехатомность Г. (см. ниже). Кроме жиров Г. всегда находится (в свободном состоянии) в вине (от 0,978%, до 1,667% — Рейхард), так как он образуется при спиртовом брожении сахара (в количестве до 3% взятого сахара — Пастер) и в очень незначительном количестве в водке (Морэн). Синтетически Г. был получен Вюрцем из трибромгидрина Г., в свою очередь образованного действием брома на йодистый аллил (см. Гликоли); Фриделем и Сильва — из трихлоргидрина, полученного взаимодействием хлористого йода с хлористым пропиленом, а так как последний был ими приготовлен присоединением хлора к пропилену, полученному из изопропилового спирта, то Г. может быть синтезирован, исходя из элементов. Г. получается, наконец, окислением аллилового спирта марганцово-калиевой солью. (Е. Вагнер — см. Глицерины). О техническом добывании Г. см. соотв. статью. Чистый Г. представляет густую, сиропообразную жидкость, обладающую сладким вкусом; он не застывает при кратковременном охлаждения до — 40°С, хотя заметно густеет; при продолжительном охлаждении до 0°, при некоторых не вполне выясненных условиях, чистый Г. способен однако закристаллизовываться, образуя расплывающиеся на воздухе ромбические кристаллы, температура плавления кот. + 17°С (по Геннингеру), + 20°С (по Нитше) и + 22,6°С (по Крауту). Уд. вес Г. — d 15/4 = 1,2637; (Менделеев, 1861, см. далее), d 20/4 = 1,2590 (Брюль и др.); Г. оптически недеятелен; показатель преломления для линии ß водорода = 1,478 (Брюль); теплоемкость = 0,612 (Винкельман). Г. смешивается во всех отношениях с водой и спиртом, но почти не растворим в серном эфире и хлороформе. Г. способен растворять в значительном количестве едкие щелочи, окиси кальция, стронция и бария, сернокислое кали, сернокислый натр, медный купорос и многие другие соли; в его присутствии хлорное железо не осаждается едкой щелочью. Под уменьшенным давлением, или с парами воды, Г. перегоняется без изменения; под обыкновенным давлением он кипит при + 20°С (Менделеев); присутствие минеральных солей, особенно же веществ, способных отнимать воду, как, напр., кислой серно-калиевой соли, фосфорного ангидрида — вызывает разложение Г., сопровождаемое потерей им воды и образованием акролеина —
С 3 Н 8 О 3 — 2Н 2 О = С 3 Н 4 О
— (Редтенбахер, Гейтер и Картмель), всегда наблюдаемого и при нагревания естественных жиров (пригорелый жир обязан своим запахом акролеину), чем последние легко отличаются от минеральных масел. При осторожном окисления (действие кислорода воздуха в присутствии платиновой черни — Гримо, или паров брома на глицерат свинца — Фишер и Тафель) Г. дает альдегид — глицерозу СН 2 (ОН).СН(ОН).СОН (см. Гидраты углерода и Глюкозы) и вместе с ней диоксиацетон СH 2 (ОН).СО.СН 2 (ОН) (Фишер). Глицероза получается и при действии азотной кислоты на Г., но кроме нее, как продукты дальнейшего окисления, образуются: глицериновая — СН 2 (ОН).СН(ОН).СООН, щавелевая — СООН.СООН, муравьиная — H.СООН, гликолевая — СН 2 (ОН).СООН и глиоксилевая — СНО.СООН кислоты; одновременно наблюдается появление синильной кислоты (Пржибытек), образование которой и объясняет получение, кроме упомянутых веществ — еще виноградной (Гейнц) и мезовинной кислот (Пржибытек, см. Винная кислота):
СН 2 (ОН).СН(ОН).СОН + HCN = СН 2 (ОН).СН(ОН).CH(OH).CN
и
СН 2 (ОН).СН(ОН).СН(ОН).CH + 2Н 2 О + О 2 = СООН.СН(ОH).СH(ОН).СООН + NH 3.
При окислении марганцово-калиевой солью из Г. может быть получена тартроновая кисл. СООН.СН(ОН).СООН (Задтлер, Пржибытек, Кампани и Биццари), но при действии избытка окислителя реакция протекает по уравнению:
C3H3O3 + C2H2O4 (щавелевая к.) + СO 2 + 3Н 2 О
(Ванклин и Фокс — способ количественного определения Г.). При нагревании с твердым едким кали Г. дает акриловую к. (Редтенбахер), при плавлении с ним — продукты ее распадения: муравьиную и уксусную кислоты (Дюма и Стас). Подобно винному спирту Г. легко реагирует с неорганическими кислотами; образующиеся продукты по химическому характеру вполне отвечают соединениям, получающимся в тех же условиях из этилового алкоголя; так, серная кислота дает глицериносерную кислоту — СН 2 (ОН).СН(ОН).СН 2.О.SО 2.ОН (получена Пелузом при растворении одной части Г. в 2-х частях серной кислоты), полный аналог серно-винной кисл. СН 3 СН 2 О.SО 2.ОН; кроме нее для Г. возможны еще две кислоты, а именно: CH 2(OH).CH(OSO2OH).CH2(OSO2 OH) и СН 2 (ОSО 2 ОН).СH(ОSО 2 ОН).CH 2(OSO2 OH); обе получены Клессоном: последняя при действии первого хлорангидрида серной кисл. — SO2 (OH)Cl на Г., а первая — при действии на глицеринотрисерную кисл. воды. С фосфорной кисл. Г. образует глицеринофосфорную кисл. — СН 2 (ОН).СН(ОН).СН 2 О.РО(ОН) 2, из сложных эфиров которой особенно замечательны лецитины — вещества, находимые в мозге, в яичном желтке, в сперматозоидах, в дрожжах, спорах и т. д. и которые при нагревании с баритовой водой распадаются на жирные кислоты (олеиновую, пальмитиновую, стеариновую и др.), глицеринофосфорную кислоту и нейрин (см. Виниловые соединения) и потому, вероятно, представляют эфиры глицеринофосфорной соли нейрина состава — СН 2 (OR').СH(ОR').СН 2 О.РО(ОН)[О.N(СН 3)3.С 2 Н 4 (ОН)], где R' остаток жирной кислоты. Нагретый с бурой, Г. дает производное борно-глицериновой кисл. (глицериноборат), обладающее кислой реакцией и имеющее довольно обширное применение в фармации как антисептическое средство. (эфиры азотной кисл. — см. Нитроглицерин; эфиры жирных и других органических кислот см. Жиры и ниже о строении Г.). Для Г. известны многочисленные галоидгидрины, которые получаются большей частью или при действии галоидоводородных кислот, или галоидных соединений фосфора на Г.; йодистый фосфор дает однако не трийодгидрин, как можно было бы ожидать, а вещество, содержащее двумя атомами йода менее — именно йодистый аллил —
CH2:CH.CH2J = CH2J.CHJ.CH2J. — J2 = C3H2J
(Бертело и Люка), жидкость, кипящую при 100 — 102°, образующую при действии йодистого водорода вторичный йодистый пропил:
СН 2:СН.CHJ + 2HJ = СН 2.CHJ. СН 3 + J2,
а при действии брома — трибромгидрин Г.
СH 2:CH.CH2J + 2Br2 = СН 2 Вr.СНВr.СН 2 Вr + JBr
(Вюрц). Подобно обыкновенному спирту, образующему алкоголяты, Г. дает при действии щелочных металлов или окисей щелочноземельных и тяжелых металлов — глицераты, большей частью кристаллические, легко изменчивые соединения; мононатрий глицерат C3H7O2 (ONa), распадаясь при нагревания, образует в значительном количестве пропиленгликоль СН 3.СH(ОH).СН 2 (ОН) (Белогубек, Лебиш и Лоос), одновременно с которым найдены еще метиловый, этиловый и нормальный пропиловый спирты, гексилен и т. д. (Фернбах). Глицерат свинца, упомянутый выше, получается при осаждения обыкновенным спиртом раствора окиси свинца в кипящем Г. При действии на глицераты галоидгидринов спиртов получен целый ряд смешанных эфиров Г.; это все жидкости, кипящие ниже Г. и напоминающие по свойствам смешанные эфиры одноатомных спиртов. Подобно гликолям, Г. способен, выделяя воду из одной частицы, давать эфироподобные ангидриды, из которых известен глицид — CH2. СН.СН 2 (СОН) — (окись аллилового спирта — Каблуков), полученный впервые Гегерфельдом; Ганрио получил глицид при действии безводного барита на раствор монохлоргидрина Г. в безводном эфире; это легкоподвижная жидкость, кипящая при 157° — 160°, смешивающаяся во всех отношениях с водой, спиртом и эфиром и быстро соединяющаяся с водой, образуя обратно Г., а при недостаточном количестве воды, — полиглицериновые спирты, вполне аналогичные полиэтиленовым алкоголям (см. Гликоли). Хлоргидрин глицида есть эпихлоргидрин (см.). Г., растворенный в воде, в присутствии мела и казеина, способен приходить в брожение, образуя этиловый алкоголь, масляную кислоту и некоторые другие продукты (Бертело, Бешан). Под влиянием брожения, вызываемого развитием Bacilus subtilis (Cohn), он образует преимущественно этильный спирт (Фиц), а под влиянием Butyl-Bacilus он дает нормальный бутиловый спирт (см.) — СН 3.СН 2.СН 2.СН 2 (ОН) (Фиц) и триметиленгликоль — СН 2 (ОН).СН 2.СН 2 (ОН) (Фрейнд, см. Гликоли). Строение Г., как двупервично-вторичного спирта — С 3 Н 8 О 3 = СН 2 (ОН).СН(ОН).СН 2 (ОН) выводится на основании следующих соображений. Выше упомянуто, что Шеврель первый указал на близость в химическом отношении жиров и эфиров органических кислот. "В самом деле, — говорит Бертело (1854), — в жирах, соединениях нейтральных, свойства кислот массированы совершенно так же, как и в эфирах, образующихся, с выделением элементов воды, при взаимодействии кислот со спиртами; оба класса соединений регенерируют кислоты и спирты, фиксируя обратно элементы воды, или под влиянием щелочей, или кислот, или воды (реакция идет быстро при + 220°С и медленно при обыкновенной темп.) с той лишь разницей, что из эфиров получаются к. и спирты, а из жиров — к. и Г.; при действии аммиака и из жиров, и из эфиров получаются амиды кислот. Еще убедительнее доказывается химическая равноценность спирта (обыкновенного) и Г. тем, что нагреванием (сложных) эфиров спирта с Г. можно, в известных условиях, получить жиры (вытеснить, следовательно, спирт из взятого эфира), обратно, нагреванием жиров со спиртом можно получить эфир спирта (и выделить в свободном состоянии Г.), а это, помимо всяких гипотез, убеждает в полной аналогии в конституции жиров и эфиров. Но если по химической натуре Г. и приближается к спирту, то, с другой стороны, формулы образуемых им с кислотами соединений и существование нескольких нейтральных продуктов для Г. и взятой кислоты — устанавливают глубокую разницу между ним и спиртом: последний дает с кислотами только один ряд нейтральных соединений, а для Г. известны три различных ряда, из которых только один по формулам отвечает эфирам спирта, так как он образуется взаимодействием одной частицы кислоты с одной частицей Г., при выделении одной же частицы воды [Бертело употребляет старые атомные веса (Н = 1, О = 8) и потому говорит 2, 4 и 6 эквивалентов воды, вместо одной, двух и трех частиц воды], второй ряд образован взаимодействием одной частицы Г. с двумя частицами кислоты при выделении 2-х частиц [Бертело употребляет старые атомные веса (Н = 1, О = 8) и потому говорит 2, 4 и 6 эквивалентов воды, вместо одной, двух и трех частиц воды] воды, и, наконец, третий, тожественный с естественными жирами, образован взаимодействием одной частицы Г. с тремя частицами кислоты, при выделении 3-х частиц [Бертело употребляет старые атомные веса (Н = 1, О = 8) и потому говорит 2, 4 и 6 эквивалентов воды, вместо одной, двух и трех частиц воды] воды... Факты эти показывают, что Г. относится к спирту, как фосфорная кислота относится к азотной кислоте", или, как говорит Вюрц (1855): "Они показывают, что Г. есть трехатомный спирт, т. е. представляет собой соединение, образованное через замещение радикалом глицерилом (C 3H5) трех атомов водорода в трех частицах воды". Правильность такого представления доказана самим же Вюрцом, как синтезом Г. (см. выше), так и получением гликолей-спиртов двухатомных (см. Гликоли), и это играло большую роль в выяснении правильного представления об атомности органических радикалов (см.). "Если этил (С 2 Н 5), — говорит Вюрц в статье о гликолях (1859), — способный соединяться с 1 эквивалентом хлора, замещает 1 эквивалент водорода [Напр., в воде, образуя винный спирт: H.O.H — вода, С 2 Н 5.О.Н — спирт; реакция осуществляется при посредстве двойного разложения: C 2H5 Cl (Вr, J, хлористый, бромистый, йодистый этил) + КОН (едкое кали) = С 2 Н 5 ОН + КCl (Вr, J, хлористый, бромистый, йодистый калий)], то этилен C 2H4, соединяющийся прямо с двумя эквивалентами хлора, подобно, напр., олову, может замещать 2 эквивалента водорода. Этилен есть двухатомный радикал, а жидкость голландских химиков — двухатомный хлорюр. Когда последний (полученный синтетически) вступает в обменное разложение с серебряной солью, то радикал остается нетронутым и становится на место 3 эквивалентов серебра. В этом и заключается теоретический интерес моего труда; им доказано, что органическая группа, соединенная с 2-мя эквивалентами хлора или брома, может, покидая их, заместить 2 эквивалента серебра. Факт этот, по-моему, нов и важен. Я попытался обобщить его, не только введя в круг исследования другие бромюры, аналогичные бромистому этилену, но и доказав, что радикал, соединенный с 3-мя атомами брома, может замещать 3 эквивалента серебра; это ясно из опыта превращения йодистого аллила в Г. Аллил, одноатомный в йодистом аллиле, делается трехатомным, поглощая три атома брома и превращаясь в трехбромистый аллил; и, введенный в реакцию с 3-мя частицами уксуснокислого серебра, он замещает три эквивалента серебра. Благодаря этим наблюдениям, учение о многоатомных радикалах, которое было неопределенной гипотезой без поддержки опытных данных, вошло окончательно в область точного знания". Первая структурная формула Г. предложена Купером (1858), а именно С 3 Н 8 О 3 = CH(OH)2.СН 2.СН 2 (ОН); А. М. Бутлеров вскоре указал, что возможна и другая формула — СН 2 (ОН).СН(ОН).СН 2 (ОН) — теперь общепринятая. Действительно, первичность двух водных остатков (гидроксилов) Г. доказывается получением из него триметиленгликоля СН 2 (ОН).СН 2.CH2 (ОН) и тартроновой кислоты СООН.СН(ОН).СООН, а содержание вторичного гидроксила явствует из того, что монохлоргидрин Г. при действии амальгамы натрия дает (Буфф) пропиленгликоль:
СН 2 Сl.СH(ОН).СН 2 (ОН) + Н 2 = СН 2.СН(ОН).СН 2 (ОН) + НCl.
К той же формуле приводит и получение Г. окислением аллилового спирта
СН 3:СН.СН 2 (ОН) + Н 2 О + О = СH 2 (ОН).СН(ОН).СН 2 (ОН)
и образование при действии йодистого фосфора на глицериновую кислоту (строение которой доказано совершенно самостоятельно — см. Г. кислота) — ß йодопропионовой кислоты —
СН 2 (ОН).СН(ОН).СООН + 3HJ = CH 2 J.СН 2.СООН + 2H 2O + J2.
Нельзя не заметить, кроме того, что это единственно возможная формула, удовлетворяющая законности Эрленмейера-Кекуле, по которой в многоатомных спиртах при одном атоме углерода может находиться не более одного гидроксила (см. Мезокзалевая кислота и Гидрат хлорала).
А. И. Горбов. Δ.
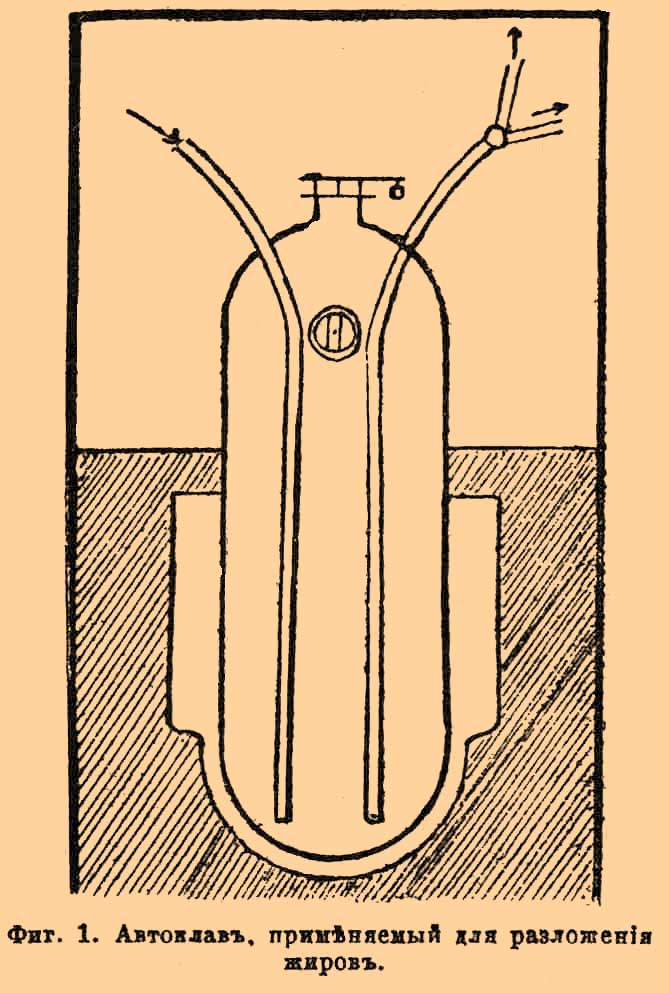
Глицерин (техн.). Г. имеет важное и обширное значение в технике. Обрабатывая смесью дымящейся азотной и крепкой серной кислот его превращают в эфир азотной кислоты С3 Н 5(NO3)2, называемый нитроглицерином, который обладает чрезвычайно сильными взрывчатыми свойствами и поэтому входит в состав различных взрывчатых веществ, особенно динамита (см.), находящих огромное применение в различных областях промышленности, особенно в горнозаводском деле. Кроме того, Г. употребляется в силу своих гигроскопических свойств в качестве прибавки к таким веществам, которые нужно предохранить от высыхания: отсюда обширное применение его в мыловаренном производстве от прибавления его к мылу, последнее долго не сохнет и остается мягким. С этой же целью он прибавляется к скульптурной глине, и ради этого же в растворе его мочат в некоторых случаях кожи при их дублении. Далее, он употребляется для сдабривания вина, пива, уксуса, ликеров и разных консервов, которым он придает сладкий вкус и предохраняет от описания. Большое количество его идет для приготовления разных косметиков, смазывания частей машин (особенно часов), приготовления протрав для некоторых красок и т. под. Общее производство Г. простирается в Европе до 3500000 пд. в год [В том числе из России 101,6 тыс. пд., на сумму 435,9 тыс. руб. (1889)], причем половина этого количества идет на приготовление динамита. Г. получается в технике исключительно как побочный продукт разложения естественных жиров и масел, производимого на стеариновых в мыловаренных зав. Естественные жиры (см.), представляя глицериды или глицериновые эфиры различных жирных кислот, при известных условиях способны разлагаться с возрождением Г. и свободных жирных кислот, которые издавна имеют обширное применение в промышленности, как материал для приготовления свеч и мыла. Упомянутое разложение в технике производится тремя способами: щелочным обмыливанием жиров, действием на них серной кислоты и разложением с перегретым водяным паром. Все эти способы имеют одинаково важное значение. Разложение жиров обмыливанием производится следующим образом. В особый, герметически закрывающийся медный котел, цилиндрической формы, так называемый автоклав (фиг. 1), вносят 2000 кг жира (бараньего или говяжьего сала или пальмового масла), 1000 кг воды и известкового молока, с таким расчетом, чтобы приходилось от 2 — 3% извести по отношению к взятому количеству жира, т. е. 40 — 60 кг на приведенную пропорцию.
Фиг. 1. Автоклав, применяемый для разложения жиров.
Через особую трубку, доходящую до дна автоклава, вводят в последний пар под давлением сначала в 4 атмосферы, которое затем увеличивают до 8 атмосфер и поддерживают последнее в течении 8 часов, после чего разложение жира окончено. Оно происходит в силу того, что сначала известь действует на жир, омыляя его, т. е. выделяет из него Г. и образует с жирными кислотами соль — мыло:
2С 2 Н 5 (RО) 3 + 3Са(НО) 2 = 2С 2 Н 5 (ОН) 2 + 3Ca(RO)2,
где RО = солеродный остаток жирной кислоты, входивший в сочетании с Г. в состав жира. Образовавшееся известковое мыло разлагается далее перегретой водой на жирную кислоту и известь:
Ca(RO)2 + H2 O = СаО + 2R(ОН).
Возродившаяся окись кальция действует на новое количество жира, обмыливает его и т. д. Когда разложение всего взятого в работу количества жира кончено, охлаждают содержимое автоклава до 130° и выпускают его через особую трубку, также доходящую до дна прибора. Трубка эта имеет кран с двойным ходом. В силу того давления, которое еще имеется в автоклаве, содержащаяся в нем жидкость вытекает из него, при открытии упомянутого крана, и, так как внизу автоклава собирается удельно более тяжелая жидкость, состоящая из раствора Г. в воде, то сначала вытекает из него последняя. Когда в трубке покажется струя жирных кислот, кран повертывают так, чтобы жидкость из него вытекала в другой приемник. Полученный раствор Г. в воде (в 5°Б.) подвергается дальнейшей обработке, с целью выделения из него чистого Г.
Разложение жиров серной кислотой ведется иначе и основано на другом процессе, а именно: если подействовать на жир крепкой серной кислотой, то последняя разлагает его с выделением свободной жирной кислоты и образованием сочетанного соединения Г. с серной кислотой, т. е. сульфоглицериновой кислоты:
С 3 Н 5 (С 18H35O2)3 + SO4H2 + 2Н 2 О = 3С 18H36O2 + С 3 Н 5 (НО) 2 SО 3 НО.
Сульфоглицериновая кислота в свою очередь легко распадается при действии воды в 100° на Г. и серную кислоту. Обмыливание жиров по этому способу в особенности приложимо к продуктам дурного качества, испорченным, прогорклым и т. п. и ведется следующим образом. Взятый жир предварительно сплавляется и оставляется в расплавленном состоянии на некоторое время, чтобы из него осели разные примеси. После этого он перепускается в железный, выложенный внутри свинцом котел с двойным дном, в котором к нему прибавляют от 6 — 12% серной кислоты в 66°Б. (12% при нечистом, смешанном жире, а 6% при пальмовом масле), при постоянном помешивании. Котел в это время нагревают, выпуская перегретый пар в пространство между стенками его дна, пока содержимое не примет температуру в 177° (на заводе Рriсе в Лондоне) или 115° (Gentilly в Париже). От действия серной кислоты сначала разлагаются, обугливаясь, различные примеси к жиру: обрывки тканей животного и т. под., отчего масса чернеет и вспучивается, от выделения сернистой кислоты; и только затем происходит образование сульфоглицериновой кислоты. Когда обмыливание кончено, массе дают 3 — 4 часа охладиться и ее перепускают в выложенные свинцом чаны, наполненные до трети водой. На дне чана находится паропроводная трубка; пропускаемым по ней паром нагревают содержимое чана до 100°, при чем сульфокислоты разлагаются, а выделенные кислоты всплывают на верх. Их сцеживают и подвергают дальнейшей очистке, а водную жидкость, содержащую Г. и серную кислоту, нейтрализуют известью и употребляют для выделения Г. Этот способ обмыливания жиров, иногда неизбежный для свечных фабрикантов, вследствие имеющегося запаса дурного жира, с точки зрения производства Г. невыгоден, так как при нем происходит значительная потеря последнего. Разложение жиров перегретым паром, т. е. водой, происходит всего лучше при температуре около 300°. Продуктами его являются только свободные жирные кислоты и глицерин. Этот способ применяется часто в Англии, напр. на большом заводе Price в Лондоне, где разложение ведется следующим образом. Сплавленный (струей пара) в резервуаре H (фиг. 2), жир пропускается, с некоторым количеством воды, в кубе Т, который нагревается снаружи топкой, пока температура не достигает 300°.
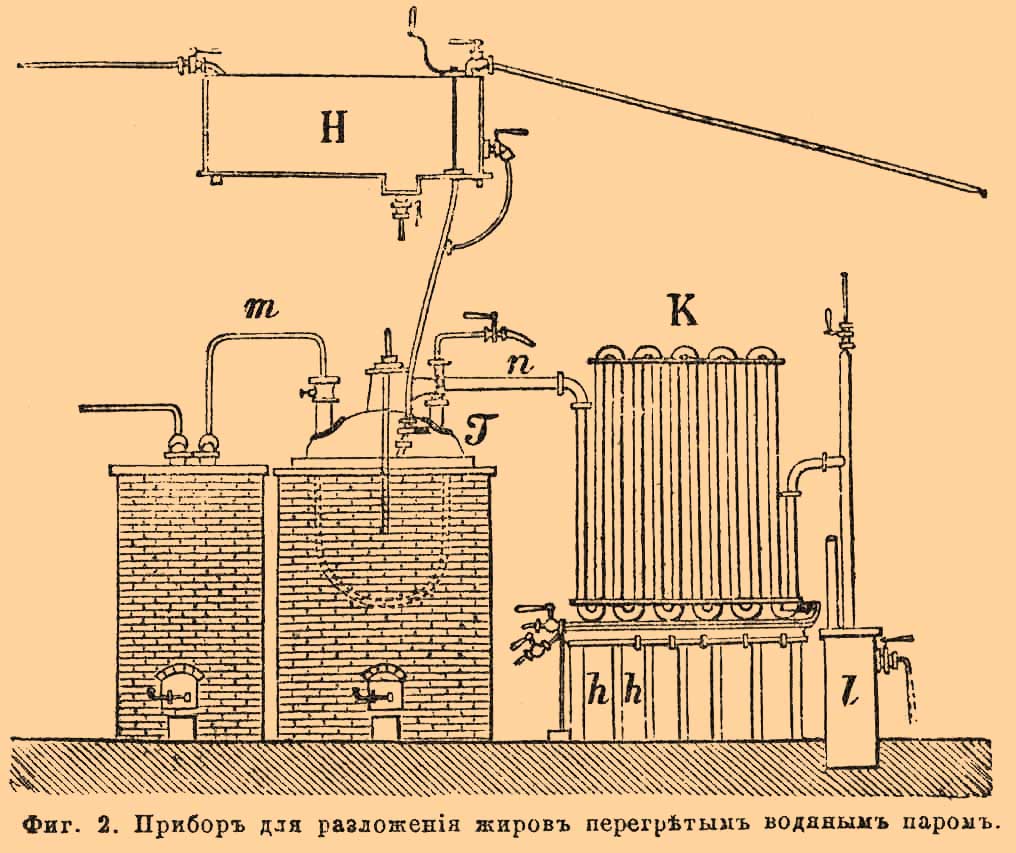
Фиг. 2. Прибор для разложения жиров перегретым водяным паром.
В это время в куб впускают непрерывно струю перегретого водяного пара по трубе m, при чем происходит разложение жира. Смесь паров воды, Г. и жирных кислот по трубке n удаляется из куба и поступает в холодильник К, состоящий из ряда вертикальных трубок, соединяющихся между собой. В изогнутом, нижнем конце каждой трубки холодильника имеется отверстие с краном, сообщающее ее с приемником h, в котором собираются сгустившиеся в холодильнике пары. Ближе к аппарату собираются почти совершенно чистые жирные кислоты, а по мере удаления от него — вместе с водой и Г. В последнем приемнике l находится уже почти одна вода. Отделенная от жирных кислот, водная жидкость, содержащая Г., перерабатывается для выделения последнего.
Выделение Г. из его водного раствора, получающегося при том или другом способе разложения жира, производится таким образом, что раствор сначала подвергается сгущению выпариванием, до удельного веса 1,12 — 1,22, всего лучше в вакуум-аппаратах, т. е. в пустоте, для чего может служить, напр., прибор, изображенный на фиг. 3. Нагревание в нем производится паром, входящим по трубе А, имеющей внутри котла тарелкообразные расширения В, B, B, увеличивающие поверхность нагревания. Паропроводная труба может вращаться вокруг своей оси, что еще более усиливает испарение. Выкачивание воздуха и паров воды производится через трубу С. Сгущенный таким образом Г. нечист и окрашен в бурый цвет. Для очищения его фильтруют через костяной уголь и подвергают вторичной перегонке, которая ведется в медных кубах, лучше всего перегретым паром и по возможности при низкой температуре, не выше 210°, а для химически чистого Г. — даже при 171°. Фильтрование через уголь и перегонку повторяют несколько раз, пока не получат продукта желаемой чистоты.
Г., как сказано выше, способен, при известных условиях, кристаллизоваться. Этим свойством часто пользуются для его очищения (на заводе Sarg в Вене и у Крестовникова в Казани). В охлажденный до 0° раствор Г. вносят несколько кристаллов ранее полученного в твердом виде Г. По истечении некоторого времени жидкость застывает в кристаллическую массу, которую помещают в центрифуги (см. Выжимание) и в последних отделяют из нее твердую часть от жидкой.
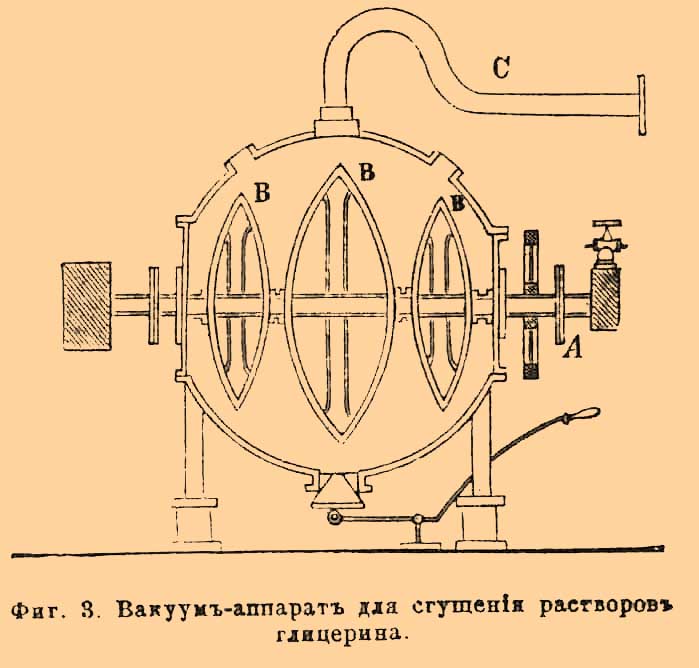
Фиг. 3. Вакуум-аппарат для сгущения раствора глицерина.
По этому способу получают весьма чистый Г. удельного веса 1,24, который дальнейшим сгущением в вакуум-аппаратах может быть доведен до 1,266. Водный раствор Г., получающийся при обмыливании жиров, с целью выделения из последних жирных кислот для фабрикации свеч, служит в настоящее время главнейшим источником для получения этого вещества, но запрос на него с каждым годом увеличивается и потому очень важно найти новый источник для его получения. В этом отношении самое важное значение имеет та водная жидкость, которая остается после приготовления из жиров (в особенности из содержащих триолеин) мыла и в которой находится большое количество Г. Но, к сожалению, до сих пор все попытки удобного и дешевого выделения из нее Г. не увенчались полным успехом, что обусловливается тем, что в такой жидкости находится, кроме Г., множество других веществ, попадающих в нее при фабрикация мыла, главным образом минеральных солей, отделение от которых Г. сопряжено с большими затруднениями. Лучший способ для этого был предложен Domeier С°, который и применяется ныне почти всюду. Domeier сначала прибавляет к жидкости до 1,5% извести, чтобы превратить находящиеся соли отчасти в едкие щелочи и выпаривает жидкость до тех пор, пока не начнут кристаллизоваться бывшие в ней соли. В тоже время образовавшиеся едкие щелочи обмыливают смолистые вещества, превращая их в смоляные мыла, которые собираются на поверхности в виде пены, увлекая с собой и остатки настоящего мыла, бывшие в жидкости. Выпаривание жидкости составляет важнейшую часть всей операции и его необходимо вести осторожно для того, чтобы выделяющиеся соли не пригорали ко дну и не производили вспучивания. Для этого, во всяком случае, нагревание должно производить сбоку прибора, а не со дна. В недавнее время Glaser предложил с этой целью особый аппарат, получивший уже некоторое распространение (фиг. 4).
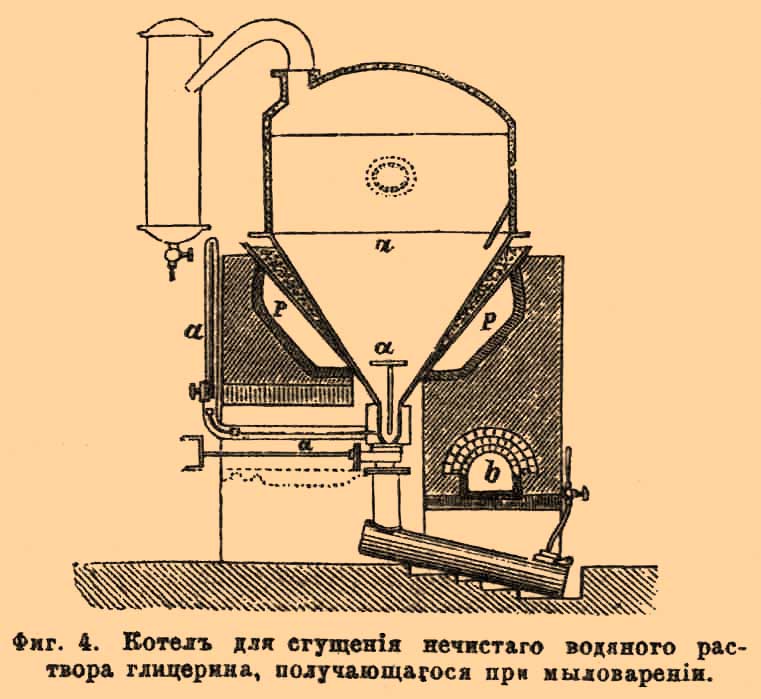
Фиг. 4. Котел для сгущения нечистого водяного раствора глицерина, получающегося при мыловарении.
Испарительный котел а имеет коническую форму и состоит из двух частей: собственно котла а и двух резервуаров, отделяющихся от котла особой перегородкой, которую можно по желанию задвигать и отодвигать, установляя в последнем случае сообщение между котлом и резервуарами. Назначение последних состоит в том, что в них собирается осадок выделяющихся при выпаривании солей. Когда последних соберется достаточно много, задвижка одного из резервуаров задвигается, резервуар таким образом отделяется от испарительного котла и может быть очищен, не прерывая всего хода испарения, которое ведется частью перегретым паром. вступающим в котел по трубке о, частью голым огнем, для чего котел а окружен с боков песчаной баней p, которая и нагревается непосредственно дымогарными газами, полученными из топки b. Полученный (остающийся в котле) сырой Г., содержащий еще до 10% минеральных веществ, подвергается, для очищения, перегонке. В некоторых случаях его предварительно очищают взбалтыванием с петролейным эфиром, сероуглеродом и т. п., для освобождения от разных жирных и смолистых примесей. Перегонка такого Г. ведется несколько отлично от того, как было описано выше. Куб берется такой же, обыкновенный; разница существует только в устройстве холодильника, который делается вроде употребляемых при перегонке и ректификации спирта. Он состоит из колонны А, А. (фиг. 5), в которой находятся перегородки — так называемые тарелки D D, пробитые мелкими (1/10 дюйма) отверстиями.
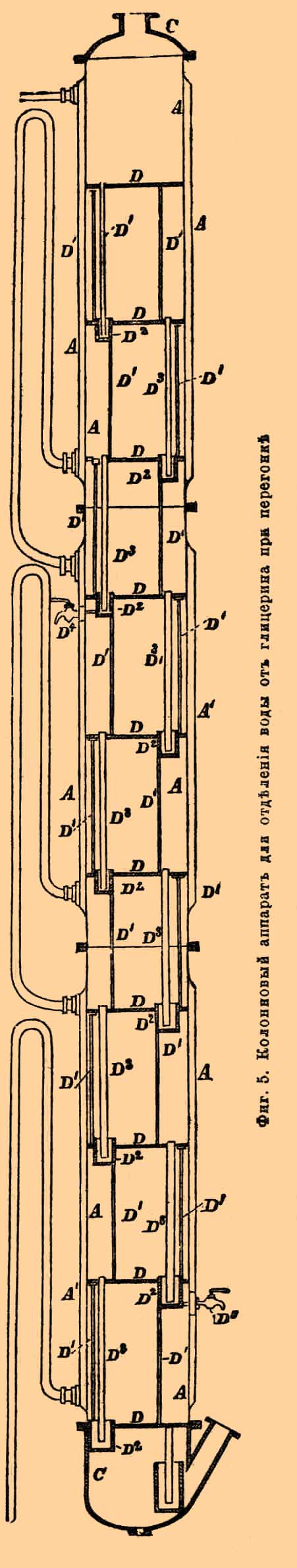
Фиг. 5. Колонновый аппарат для отделения воды от глицерина при перегонке.
Пары Г. и воды, поднимающиеся снизу от С, идут вверх, сгущаются на упомянутых тарелках и стекают по трубкам D, в приемники D2, снабженные кранами D1. Внизу колонны, на ближайших тарелках, сгущается почти чистый Г., а на более удаленных — смешанный с водой. Такой водный Г. подвергается дальнейшей перегонке обычным путем. Полученный описанными способами, Г. поступает в продажу в различном состоянии чистоты, почему необходимо при покупке его обращать внимание на последнюю, так как во многих применениях Г. требуется совершенно чистый продукт. Обыкновенно в продаже встречаются следующие сорта: 1) сырой Г.; 2) химически чистый Г., который однако же содержит еще часто 6 — 10% воды. Он не должен при нагревании с серной кислотой и спиртом давать никакого эфирного запаха, а с уксусно-известковой солью — осадка; 3) динамитный Г. в 28°Б. желтоватого цвета; он должен быть свободен от извести и давать с раствором ляписа только чуть заметную муть; 4) белый Г., не содержащий извести, но менее чистый, чем предыдущий; 5) желтый Г., также не содержащий извести. Присутствие во всех этих сортах разных посторонних веществ органического происхождения лучше всего узнается помощью раствора свинцового уксуса (основной уксусно-свинцовой соли), который в этом случае производит в Г. более или менее обильный осадок. Если последний получается — Г. негоден для приготовления динамита. Из числа органических веществ жирные кислоты, жиры и смолы могут быть определены взбалтыванием испытуемого Г. с хлороформом, который будет извлекать все подобные вещества. Испаряя затем отделенный от Г. слой хлороформа, можно получить в твердом виде остаток этих веществ. Присутствие извести открывается тем, что к водному раствору испытуемого Г. прибавляют раствора щавелево-аммиачной соли; в случае нахождения извести получается белый осадок. Все же вообще минеральные вещества могут быть определены сжиганием определенного (5 г) количества Г. Минеральные вещества будут в остатке. Количество их в хороших сортах не должно превышать 0,1%, самое большее 0,2. Хороший Г. должен смешиваться со спиртом, образуя совершенно прозрачный раствор, не должен чернеть при нагревании с крепкой серной кислотой и при смешивании с 10% раствором ляписа не должен выделять, даже при стоянии (в темном месте), черного осадка восстановленного серебра.
Ср. S. Koppe, "Das Glycerin, seine Darstelung n. Anwendung" (1889). О разложении жиров см. сочинения по приготовлению свеч и мыла при стеарине.
И. И. Канонников. Δ.
Глицерин (способ определения). Г. открывается легко только в водных растворах. Качественными реакциями на него служат: характерный запах акролеина, образующегося при выпаривании досуха раствора с кислым сернокислым кали и нагревании затем остатка (выделяющиеся газы можно собрать в воде и доказать образование акролеина помощью фуксино-сернистой кислоты [Окрашивание в розовый цвет бесцветных водных растворов фуксино-сернистой кислоты при прибавлении исследуемого вещества служит характерной реакцией для всех одноатомных альдегидов (Шифф и Каро), позволяющей отличать их от кетонов, которые этой реакции не показывают. При описываемой пробе на Г. окрашивание появляется довольно медленно. достигая наибольшей яркости минут через 15— 20 стояния. Опыт ведут на холоду, так как раствор фуксино-сернистой кислоты окрашивается от нагревания. Маннит, тростниковый сахар, глюкоза, молочный сахар, крахмал, декстрин, желатина, кислоты стеариновые и олеиновые не дают окрашивания, но присутствие гидратов углерода в Г. уменьшает чувствительность реакции, потому что продукты, образующиеся при их нагревании с кислой серно-калиевой солью, мешают появлению розового окрашивания, и в случае присутствия углеводов должно начать с их удаления (см. ниже определение Г.). При открытии Г. в молоке предварительно удаляют казеин, альбумин и молочный сахар. (Кон)]; появление зеленого цвета при внесении в пламя горелки кусочка буры, смоченного раствором Г. (предварительно должно быть доказано отсутствие аммиачных солей, или же они должны быть удалены) и карминово-красное окрашивание при прибавлении аммиака к охлажденному раствору Г., нагретому ранее до 120° с серной кислотой (Рейхль). Количественное содержание Г. в водных растворах можно вычислить на основании удельного веса или показателя преломления раствора (числовые данные см. ниже); но для этого необходимо, чтобы раствор был заведомо чист [По требованиям германской фармакопеи чистый Г. не должен ни давать серебряного зеркала, ни окрашиваться в желтый цвет (в течение пяти минут), при следующей пробе: раствор равных частей Г. и водного аммиака нагревают до кипения при постоянном помешивании, затем пламя удаляют и прибавляют несколько капель аммиачного раствора азотнокислого серебра; проба, главным образом, направлена к открытию содержания мышьяковистого ангидрида в Г., но она совершенно не удается — даже при заведомом содержании нескольких % Аs 2 О 3 — в случае избытка аммиака (Яффе, Лютке). Присутствие же акролеина в Г. легче определяется по запаху, или по розовому окрашиванию с раствором фуксино-сернистой кислоты (Лютке), или по получению бурого (красного) осадка при взбалтывании с реактивом Несслера (щелочной раствор двойной соли йодистого калия и йодистой ртути); осадок, образующийся с альдегидами, отличается от осадка, даваемого реактивом с аммиаком (см.), тем, что с цианистым калием он чернеет, между тем, как осадок от аммиака исчезает от прибавления цианистого калия, реакция в высшей степени чувствительная (Кризмер)]. Из методов определения содержания Г. в продажных образцах его отметим следующие: Шампион и Пелле определяют Г. в виде нитроглицерина, для чего вещество обрабатывают смесью концентрированных азотной и серной кислот (Дикман советует прибавлять при охлаждении по каплям смесь 5 ч. серной кислоты в 66°Б с 3 ч. азотной кислоты в 48°Б.; серная и азотная кислоты не должны содержать соляной кислоты и в азотной кислоте может быть не более 1% азотистой кислоты); образовавшийся нитроглицерин промывают водой, растворившуюся в промывных водах часть извлекают эфиром, сушат до постоянного веса на водяной бане и взвешивают; 190 ч. нитроглицерина отвечают 100 ч. Г. Безопаснее способ Моравского, состоящий в том, что 1 ч . сырого Г. смешивают с 25 ч. окиси свинца, выпаривают и сушат до постоянного веса при 130°. Привес окиси свинца отвечает всем нелетучим примесям, содержавшимся в Г., и его остатку С 3 Н 6 О 2; количество нелетучих примесей определяют (приблизительно) высушиванием навески Г. до постоянного веса при 160° (Г. при этой температуре довольно быстро испаряется), вычитают из привеса окиси свинца и разность умножают на 1,3429. Способ удобный, но применимость его ограниченная, так как он дает хорошие результаты только с довольно чистыми образцами Г., содержащими кроме хлористого натрия лишь незначительные количества посторонних органических веществ; когда же имеется подмесь сернокислых солей, свободных щелочей или смолистых веществ, то не удается вполне удалить Г. нагреванием при 16 0 ° и, кроме того, невозможно (без более сложных приборов) избежать поглощения углекислоты едкими щелочами (Генер). По Бенедикту и Кантору, кипятят (в продолжение 1 часа) Г. (1 г) с уксусным ангидридом (7 г) и безводной уксусно-натровой солью (3 г) в колбе с обратно поставленным холодильником (иначе происходит потери вследствие значительной летучести триацетина с парами воды); затем раствор охлаждают; прибавляют 50 куб. см воды; слегка подогревают, чтобы облегчить растворение осевшего масла; отфильтровывают от белого хлопьевидного осадка, который содержит большую часть органических примесей Г. и может быть очень значительным; снова охлаждают; усредняют в присутствии фенолфталеина уксусную кислоту слабым раствором едкого натра, избегая его избытка; омыляют триацетин раствором едкого натра (приблизительно 10%) и титруют обратно его избыток нормальной соляной кислотой, которой точно определяют и титр едкого натра, употребленного для омыления; разность обоях титрований представляет количество соляной кислоты, пошедшей на омыление триацетина; 1 куб. см нормальной соляной кислоты отвечает 0,03067 г Г. Вследствие легкой омыляемости триацетина, даже при строгом соблюдении приведенных условий, получаются по этому способу большей частью слишком низкие цифры; он совершенно неприменим, если исследуемый раствор содержит менее 30% Г. (Генер). Вполне по принципу аналогичен более простой способ Баумана-Дица, состоящий в том, что раствор Г. (около 0,1 г Г. в 10 — 20 куб. см воды) взбалтывается 10 — 15 минут с хлористым бензоилом (5 куб. см) и едким натром (35 куб. см 10 % раствора), выделяющийся осадок растирается со щелочной жидкостью (для полноты удаления хлористого бензоила) и, после кратковременного стояния, собирается на высушенном при 100°С фильтре, промывается водою и, наконец, сушится 2 — 3 часа при 100°С 3,85 ч. полученной смеси ди- и трибензоата [Смесь эта плавится, после перекристаллизации из эфира, довольно постоянно при + 170°С и образование ее в указанных условиях может служить и качественной реакцией на Г. При употреблении раствора едкого натра большей крепости получается исключительно трибензоат (Диц, Панормов)]. отвечают 1 ч. Г. Другие многоатомные спирты и углеводы, способные также с хлористым бензоилом давать сложные эфиры, должны отсутствовать, или же их необходимо предварительно удалить [Кроме приведенных способов, на которых наиболее просты первый и последний, описано и рекомендовано еще много других, состоящих в окислении Г. и количественном определении какого-нибудь из образующихся при этом продуктов]. Это особенно важно при определении содержания Г. в пиве и вине, где поступают следующим образом: 50 куб. см пива выпаривают досуха с примесью песка и известкового молока; остаток мелко растирают, извлекают 50 куб. см 96% спирта, к охлажденной вытяжке прибавляют 75 куб. см абсолютного эфира, который осаждает мальтозу и парапептон; фильтрат выпаривают на водяной бане; остаток сушат при 100° — 150°С, растворяют в 5 — 10 куб. см воды и взбалтывают с 2 — 3 куб. см хлористого бензоила и 7 ч. 10% едкого натра; затем поступают по вышеуказанному. Точно так же анализируются и вина, с той лишь разницей, что при перебродивших, бедных сахаром белых и красных винах выпаривают с известковым молоком 20 куб. см вина, остаток извлекают 20 куб. см 96% спирта, по осаждении прибавляют 30 куб. см безводного эфира, фильтруют, осадок промывают смесью спирта (2 ч.) с эфиром (3 ч.) и фильтрат выпаривают на водяной бане, а при сладких винах к 20 куб. см вина кроме известкового молока прибавляют еще 1 г песка, а количества спирта и эфира удваивают; для окончательного определения берут не более 0,1 — 0,2 г сырого Г., выделенного таким образом. При определении содержания Г. в жирах к 100 ч. расплавленного жира прибавляют 65 ч. кристаллического гидрата окиси бария; массу старательно растирают; прибавляют для облегчения омыления еще 80 куб. см 95% спирта; затем, когда все затвердеет, кипятят (в продолжение 1 ч.) с 1 литром воды; растертый осадок баритовых солей промывают еще раза два водой; все водяные вытяжки подкисляют серной кислотой, выпаривают на половину; избыток серной кислоты удаляют углекислым баритом; фильтрат сгущают до 50 куб. см и окончательно содержание в нем Г. устанавливают одним из описанных выше способов.
А. И. Горбов. Δ.
Удельный вес водных растворов глицерина. Чистый Г. в жидком виде при обыкновенной температуре представляет густую, сиропообразную жидкость, очень мало изменяющую свои внешние свойства от прибавки воды. Но так как от прибавления воды уд. вес уменьшается, то содержание Г. в растворе проще всего (когда нет иных подмесей) определяется с помощью удельного веса [Для той же цели применяют также определение показателя преломления света и упругости паров растворов Г.]. Определения, сделанные мной в 1861 г. для безводного глицерина, согласуются с позднейшими более подробными исследованиями В. Ленца (1880) и Герлаха (1884) и в результате [Подробности см. Менделеев, "Исследование водных растворов по уд. весу"] получается следующая таблица, в которой p означает содержание Г. в процентах по весу, s есть удельный вес при 15°С, считая воду при 4° за 10000 [в безвоздушном пространстве], ds/dp есть изменение (производная) уд. веса при возрастании содержания Г. на 1% и ds/dt есть изменение уд. веса при возрастании температуры на 1°С.
| р = 0% | s = 9992 | ds/dp = 23,6 | ds/dt = — 1,5 |
| 10 | 10233 | 24,5 | —2,0 |
| 20 | 10473 | 25,3 | —2,2 |
| 30 | 10739 | 26,2 | —2,8 |
| 40 | 11005 | 27,0 | —3,5 |
| 50 | 11279 | 27,8 | —4,1 |
| 60 | 11562 | 28,7 | —4,6 |
| 70 | 11845 | 27,8 | —5,2 |
| 80 | 12118 | 26,9 | —5,4 |
| 90 | 12382 | 25,9 | —5,7 |
| 100 | 12637 | 25,0 | —5,7 |
Данные для уд. веса выражаются с достаточной для практики точностью двумя параболами:
От р = 0 до р = 63%: S = 9992 + 23,65р + 0,0420р 2
От р = 63% до p = 100%: S = 9671 + 34,33p — 0,0467р 2
Промежуточное же соединение (р = 63,0% Г.) отвечает составу C 3H3O3 + 3H2O.
Д. Менделеев.
|
Page was updated:Tuesday, 11-Sep-2012 18:15:04 MSK |