| [ начало ] | [ Й ] |
Йодометрия
— представляет один из изящных приемов объемного (титрования) анализа. Сюда отнесены все те приемы анализа которые так или иначе сводятся к количественному определению йода объемным путем. Вещества, анализ которых входит в круг этого рода определений, вообще говоря, можно разделить на две категории: 1) йод и те вещества, которые выделяют его из KJ или непосредственно, как, напр., хлор, бром, или косвенным путем, как, напр., К 2 СrO 4 с HJО 4 или КСlO 3 с НCl и пр.; 2) соединения, которые окисляются в присутствии йода: SO 2, Аs 2O3 и пр. Определение йода, на котором основаны все расчеты при этих исследованиях, ведется главнейшим образом при помощи серноватисто-натриевой соли Na 2S2O3, при чем индикатором служит крахмал, который, как известно, дает со свободным йодом синее окрашивание. Применение Na 2S2 О 3 основано на превращении этой соли в присутствии йода в тетратионовую соль натрия:
J2 + 2Na2S2O3 = Na2S4O6 + 2NaJ.
Реакция эта идет очень гладко и до конца. Продажная чистая соль имеет состав приблизительно Na 2S2O3.5H2 O; в случае недостаточной чистоты (от примеси хлористых металлов, сернокислых солей) ее перекристаллизовывают; она не должна содержать углекислых, сернисто-кислых солей и свободной сернистой кислоты [Необходимо иметь в виду, что для определения СО 2, Н 2SO4 в Na 2S2O3 недостаточно простого испытания раствором ВаCl 2, так как ВаСО 3, ВаSO 4 значительно растворяются в Na 2S2O3, нужно предварительно с помощью йодного раствора обратить Na 2S2O3 в Na 2S4O6 и уже потом пробовать р-ром BaCl 2, при этом, конечно, если в соли находились соединения SO 2, они тоже превратятся в сернокислые.]. На практике, при анализах, обыкновенно употребляют 1/10 нормальный раствор соли, т. е. заключающий 24,764 г Nа 2S2 О 3.5Н 2 O на 1 л (ат. в. Na = 22,99, S = 31,98, O = 15,96, J = 126,54, Н = 1); иногда применяют и 1/100 норм. раствор. Хотя существуют указания, что Na 2S2O3.5H2 O в эксикаторе над Н 2 SО 4 выделяет всю воду, и таким образом можно было бы знать титр (содержание безводной соли) приготовленного раствора, но лучше определять этот титр по йоду, тем более что последний можно иметь чрезвычайно легко в чистом виде. Для этой цели берут навеску йода (от 0,3 д. до 0,5 д.) и бросают в воду, к которой прибавлено некоторое количество KJ (для облегчения растворения, с одной стороны, и уменьшении летучести йода, с другой) и приливают исследуемого раствора Na 2S2O3 из бюретки до тех пор, пока жидкость из темно-бурой не сделается слабо-желтой; тогда прибавляют к ней крахмального раствора и, постоянно перемешивая, вновь приливают серноватисто-натриевой соли до момента исчезновения синего окрашивании. Титр раствора выражают прямо в количестве йода, израсходованного на 1 куб. см раствора. Если раствор 1/10 норм., титр его = 0,012654 г йода; в противном случае, для удобства расчетов, предпочитают раствор подогнать к этому содержанию прибавлением соответственного количества воды или соли. Нужно заметить, что растворы Na 2S2O3 при хранении с течением времени изменяются (жидкость мутится, происходит выделение серы и пр.), что обусловливается многими причинами; с одной стороны, показано участие в этом процессе углекислоты воздуха в присутствии кислорода, с другой — немалую роль играет действие прямых солнечных лучей и, вероятно, связанное с ним нагревание. Для избежания этого советуют готовить растворы на воде прокипяченной, держать их в прохладном месте и не на свету; кроме того предлагали прибавлять к раствору немного углекислого аммония или К 2 СО 3 для образования с могущей присутствовать углекислотой двууглекислой соли, имея таким образом в виду устранить ее действие на Na 2S2O3; но польза от такой прибавки сомнительна, так как в присутствии щелочей и их углекислых солей вследствие образования (вероятного) йодноватистых солей, которые не реагируют с Na 2S2O3 подобно свободному галоиду, йода всегда определяется меньше, иногда даже на много процентов (Topt, "Zeitschr. f. an. Chemie", 1887), и следовательно, титрование при такой прибавке К 2 СО 3 должно было бы вестись в кислой среде. При употреблении Na 2SO3 нужно не упускать из виду, что энергичными кислотами (НCl, H 2SO4) эта соль постепенно разлагается с выделением серы и образованием SO 2, отношение которой к йоду, как мы увидим дальше, совершенно иное; поэтому в тех случаях, когда приходится определять йод в кислой среде, необходимо приливать раствор Na 2S2O3 при энергичном помешивании титруемой жидкости; тогда взаимодействие между Na 2S2O3, а йодом наступает раньше, чем соль разложится. При самом определении титра Na 2S2O3 помощью йода существует некоторое неудобство, состоящее в том, что чистый йод при хранении с течением времени изменяется (с одной стороны, он притягивает влагу, а с другой, самое главное, — на него действует пыль, плавающая в воздухе и попадающая в склянку при откупоривании), и поэтому перед определением титра советуют подвергать йод возгонке; что же касается готовых растворов его, то они еще менее постоянны. Чтоб устранить это неудобство были предложены другие приемы, которые все основаны на титровании йода, выделенного из чистого KJ в известных условиях. Для этой цели можно пользоваться (Volhard) реакцией:
K2Cr2O7 + 6KJ + 7H2 SО 4 = 4K2SO4 + Cr2(SO4)3 + 6J + 7Н 2O.
Количество взятой К 2 Сr 2O7 определяет количество выделенного йода, который затем титруют исследуемым раствором Na 2S2O3. Так как K 2Cr2O7 легко получается в совершенно чистом виде, раствор ее хорошо сохраняется и состав раствора легко может быть определен, исходя из веса взятой соли, то, приготовляя раствор этой соли определенной крепости и беря его каждый раз по объему, определение титра Na 2S2O3 этим способом сильно можно упростить. К сожалению, реакция выделения йода из KJ, по некоторым исследованиям, требует времени от 1/4 до 1/2 часа, в особенности при слабых растворах. Кроме того, присутствие окрашенных солей окиси хрома вредит чувствительности показаний крахмала. Вместо К 2 Сr 2O7 более пригодными оказались KJO 3 и Na 2BrO3, употребляемые в растворах определенной крепости [По некоторым указаниям, растворы KJO 3 могут покрываться плесенью, почему к ним советуют прибавлять какого-либо антисептика, напр. NaCl].
КJО 3 + 3Н 2 SО 4 + 5KJ = 3К 2 SО 4 + 3J2 + 3Н 2 О.
Само собою разумеется, что KJ, применяемый в этих случаях, должен быть совершенно чист и не содержать KJO 3, как иногда случается. Примесь KJO 3 к KJ легко открыть, приливая к раствору KJ серной кислоты и пробуя жидкость крахмальным раствором. Наряду с серноватисто-натриевой солью, другим основным раствором при йодометрических исследованиях служить титрованный раствор йода, назначенный для определения веществ, окисляемых в присутствии йода. Применяют обыкновенно 1/10 нормальный раствор, т. е. содержащий 12,654 г йода на 1 литр, иногда же 1/20 нормальный и даже 1/100 норм. Готовят его простым растворением соответственного количества йода в водном растворе KJ (около 20 г соли на 1 л), а титр определяют при помощи титрованного раствора Na 2S2O3. Йодные растворы хранятся в склянках с хорошо притертыми пробками и при употреблении по возможности не приводятся в соприкосновение с обыкновенными или каучуковыми трубками и пробками (бюретки берут со стеклянными кранами и пр.). Титр раствора довольно быстро меняется, что зависит, с одной стороны, от испарения йода и с другой — от медленного взаимодействия между йодом и водой (жидкость постепенно обогащается HJ). Такое изменение идет энергичнее при освещении раствора прямыми солнечными лучами и при доступе воздуха; почему хранить эти растворы советуют в темном месте и в сосудах небольшой емкости, по доверху наполненных жидкостью. Раствор крахмала (индикатор) готовится, растирая продажный крахмал, в небольшом количестве холодной воды, потом обливая его кипящей водой (1 ч. на 100 ч. воды) и оставляя полученный мутный раствор отстаиваться в высоких цилиндрах. При титровании употребляют только прозрачную жидкость, беря всегда определенное количество, напр. 1 куб. см, и наблюдая, чтобы титруемая жидкость не была нагрета, так как при повышении температуры синее соединена крахмала с йодом распадается. Крахмальный раствор при хранении довольно быстро изменяется, и чувствительность его сильно ослабевает; для устранения этого неудобства предложено прибавлять: ZnCl 2, CaCl2, салициловую кислоту, NaCl и пр. Из них хлористый цинк ZnCl 2 имеет наибольшую консервирующую способность; но, к сожалению, он не может быть применен в тех случаях, когда при титровании могут образоваться нерастворимые цинковые соли, напр. при анализе сернистых соединений; то же можно сказать и о СаCl 2; для всех случаев анализа более всего удобен NaCl; но необходимо раствор, насыщенный этой солью, хранить в прохладном месте. Можно употреблять также так назыв. "растворимый крахмал" (Zulkowski), приготовляемый следующим образом. около 60 г хорошо измельченного крахмала (лучше всего картофельного) смешивают с 1 кг глицерина и постепенно нагревают до 190°, при чем происходит растворение и превращение его в растворимое в воде видоизменение, особенно — если держать эту температуру около 1/2 часа. Для получения этого вещества в чистом виде глицериновый раствор охлаждают до 120° и выливают тонкой струей в крепкий спирт (на 1 ч. раствора 2—3 ч. спирта), причем растворимый крахмаль выделяется в осадке; его промывают спиртом, растворяют в воде, фильтруют и вновь осаждают спиртом. Чистый препарат легко растворим в воде и слабом спирте, в закупоренных сосудах хорошо сохраняется; чувствительность его водного раствора к йоду одинакова с обыкновенным крахмальным раствором. Рассмотрим теперь применение титрованных растворов Na 2S2O3 и йода для разных случаев анализа.
Йод. Определение йода в свободном виде уже ясно из предыдущего; для определения же его в йодистых металлах требуется предварительно выделить его. Это достигается множеством способов, из которых укажем некоторые. Самое простое — нагревать йодистые металлы с солями окиси железа, например
Fe2Cl6 + 2KJ = Fe2Cl4 + 2KCl + 2J,
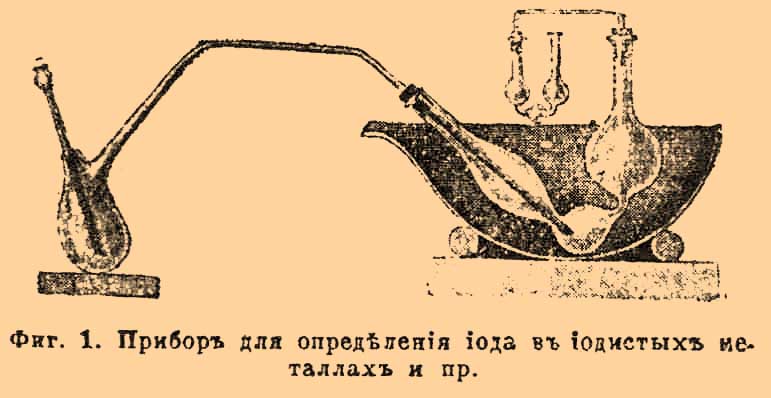
но лучше всего идет эта реакция с кислой сернокислой солью окиси железа в присутствии некоторого количества свободной Н 2 SО 4. Железной соли берут избыток и реакцию ведут при нагревании в особом баллоне с отводной трубкой; для поглощения йода служить водный раствор KJ; прибор так устроен, что через него возможно пропускать ток воздуха или СО 2 для удаления последних следов J из баллона (см. фиг. 1).
Фиг. 1. Прибор для определения йода в йодистых металлах.
Надо заметить, что этим путем не удается выделить всего J из AgJ, Cu 2J2, HgJ. Для растворимых йодистых металлов пользуются свойством меди давать соединения с йодом, отвечающие закиси, при двойном разложении между солями окиси меди и йодистым металлом, напр.
2KJ + CuSO4 = CuJ + K2SO4 + J;
при этом половина всего йода выделяется в свободном состоянии; по некоторым указаниям результаты сильно зависят от концентрации раствора и пр. Можно указать на применение окислов азота для выделения йода и проч.
Хлор, бром. Определение свободного хлора или брома, напр. в хлорной или бромной воде, делается таким образом: известное количество исследуемой жидкости смешивается с избытком раствора КJ и титруется выделенный в эквивалентном количестве йод. Множество кислородных соединений способно выделять из соляной к-ты хлор в количестве эквивалентном или всему находящемуся в них кислороду, напр. КClО 3, или известной части его, напр. К 2 Сr 2O7, смотря по их природе; на этом основано определение их. Выделившийся при разложении НCl хлор пропускают в раствор KJ, взятого в избытке (иначе могут образоваться соединения хлора с йодом) и по окончании реакции титруют выделенный йод. Расчет полученных данных не представляет затруднения, если известен ход реакции. Этим путем определяются хромовая кислота и ее соли, соли хлорноватой, бромноватой, йодноватой кислот (хотя они и прямо выделяют J из KJ в присутствии, напр., Н 2 SО 4), перекиси, напр. MnO 2, PbO2 и проч. Реакцию ведут или в приборе, изображенном на фиг. 1, или на фиг. 2.
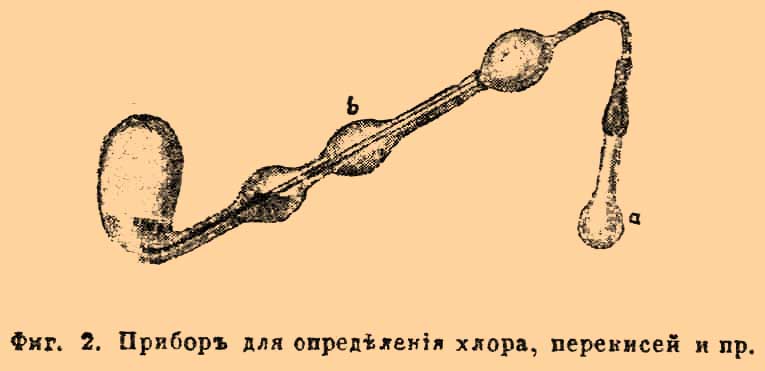
Фиг. 2. Прибор для определения хлора, перекисей и др.
Во многих случаях эта операции упрощается и заменяется непосредственным нагреванием исследуемого вещества с НCl и KJ в толстостенном стеклянном сосуде с хорошо пришлифованной пробкой, которая прочно держится на своем месте при помощи особого зажима (см. фиг. 3).

Фиг. 3. Герметически запирающаяся склянка для различных реакций при нагревании.
При разложении склянку держат известное время в нагретой воде; по окончании реакции ее охлаждают и определяют йод. Чтобы избежать окисления йода во время нагревания на счет воздуха, находящегося в склянке, перед началом операции его заменяют углекислотой (очевидно, что кислород воздуха, поглощенный йодом, в дальнейшем с НCl дает соответственное количество хлора, в конце концов убыль его выразится в выделении эквивалентного количества йода.). Подобный упрощенный ход определения различных кислородных соединений возможен только в том случае, когда рядом с главной не идут побочные реакции, выделяющие йод из KJ; например, нельзя было бы применять этот способ в присутствии железных и медных содей, выделяющих йод из KJ, но не разлагающих НCl. Вещества, окисляющиеся в присутствии йода, и анализ которых входит в круг йодометрических определений, крайне разнообразны. Сюда принадлежат SO 2 и ее соли, Н 2 S и сернистые металлы, серноватистые соли, закисные соединения различных родов, напр. для Sn, As 2O3, Sb2 О 3 и проч. Определение здесь ведется таким образом: к исследуемой жидкости, к которой прибавлено крахмального раствора, приливают титрованного раствора йода до появления фиолетового окрашивания; зная количество израсходованного йода и ход реакции окисления, мы имеем все данные для расчета.
Сернистая кислота. Определение SO 2 в слабых растворах идет довольно правильно: SO 2 в присутствии йода окисляется в серную кислоту
SО 2 + J2 + 2Н 2 О = Н 2SO4 + 2НJ.
Эта реакция идет настолько хорошо, что Бунзен предложил употреблять титрованный раствор SO 2 для определения йода при йодометрических исследованиях; для более или менее крепких растворов (уже начиная с 0,03—0,04% раствора) явление сильно усложняется, результаты титрования колеблются, и содержание SO 2 определяется ниже действительного. Это довольно долго объяснялось тем, что реакция
SO2 + J2 + 2H2O = H2SO4 + 2HJ
идет в ту и другую сторону для крепких растворов, т. е. с увеличением содержания Н 2 SО 4 происходит разложение HJ; но потом показано было (Volhard), что H 2SO4 здесь роли не играет, что в крепких растворах при избытке SO 2 происходит взаимодействие между SO 2 и HJ с выделением йода и серы, напр. реакция может идти так:
4HJ + SO2 = 4J + S + 2H2O.
Это именно и происходит при обычных условиях анализа, т. е. когда раствор йода приливают к раствору SO 2, при чем образовавшийся HJ находится в присутствии большого избытка SO 2; если же приливать SO 2 к йоду, то SO 2 целиком окисляется в Н 2 SО 4 и указанная реакция не успевает наступить; таким образом можно определять и довольно крепкие растворы SO 2.
Сероводород (H2 S) в присутствии йода разлагается с выделением серы:
Н 2S + 2J = S + 2HJ.
Определение это лучше всего вести, прибавляя, напр., к водному его раствору избыток раствора йода и определяя потом излишек йода при помощи Na 2S2O3, так как при самой реакции Н 2 S с йодом конец ее выступает не особенно резко. На таком определении Н 2 S основан способ, предложенный Клобуковым, для анализа различного рода соединений серы: сернистых металлов, сернисто-кислых, тионовых солей и проч., разлагаемых вообще соляной кислоты с образованием Н 2 SО 4, SO2, H2 S и выделением йода. По способу Клобукова, эти соединения разлагаются НCl в присутствии некоторого избытка цинка; тогда продуктом разложения являются только Н 2 SО 4 и Н 2 S; первая определяется обычным путем после окончания операции, а Н 2 S пропускается в титрованный раствор йода в KJ.
Железо. Определение железа йодометрическим путем было предметом многих исследований. Хотя обычный прием анализа железных соединений помощью хамелеона дает прекрасные результаты, но здесь необходимо иметь очень чистый цинк для восстановления окисных соединений железа в закисные; кроме того сама операция требует продолжительного времени; между тем, при йодометрическом методе определение ведется с окисными соединениями железа, что чаще всего встречается на практике, и это несколько упрощает дело. Мор предложил для этой цели воспользоваться реакцией выделения йода солями окиси железа из KJ. Опыт ведется обыкновенно с Fe 2Cl6 в присутствии НCl при нагревании до 50° — 60° в закрытом сосуде в условиях, как указано выше для выделения йода. Полнота разложения железной соли при этой реакции зависит от концентрации ее раствора, и хотя нагревание способствует этому, однако, при титровании выделившегося здесь йода до обесцвечивания необходимо наблюдать, не появится ли вновь синее окрашивание при новом нагревании — признак, что осталась еще железная соль неразложенная. Если это случается, продолжают операцию, пока это не перестанет повторяться. Для уменьшения ошибок, могущих быть при всех этих операциях, лучше всего титр Na 2S2O3 здесь выразить непосредственно в железе. Для этой цели берут навеску чистой железной проволоки (1 г) растворяют осторожно в НCl (стараясь, чтобы не было потерь жидкости от выделения газа) и окисляют FeСl 2 в Fе 2Cl6 при помощи КClО 3 или лучше Н 2O2; после этого, разбавивши раствор до известного объема (позаботившись, конечно, о полном разрушении прибавленного окислителя и об удалении при помощи нагревания следов выделившегося хлора), берут определенное количество раствора и проделывают с ним всю вышеуказанную операцию определения железа. Зная количество взятого для опыта железа, определяют сколько приходится его на 1 куб. см израсходованного Na 2S2O3. Раствор Fе 2Cl6 хорошо сохраняется и раз приготовленный долго может служить для вышеозначенной цели. Фрезениус предложил вести определение железа при помощи хлористого олова, SnCl 2, которое, как известно, восстановляет окисные соединения железа в закисные:
Fe2Cl6 + SnCl2 = 2FeCl2 + SnCl4.
Скорость этой реакции зависит от концентрации раствора железной соли и температуры. По Фрезениусу, SnCl 2 прибавляется в некотором избытке и потом обратно титруется раствором йода. Опыт ведется таким образом: раствор железной соли (обыкновенно ее имеют в виде Fe 2 Сl 6) в присутствии НCl нагревают до кипения и к нему прибавляют SnCl 2 из бюретки до тех пор, пока жидкость, окрашенная в желтый цвет, не станет бесцветной, стараясь избегать большого избытка SnCl 2. После того жидкость охлаждают, устраняя доступ воздуха, в присутствии которого SnCl 2 окисляется в SnCl 4 (для этого, нагрев до кипения, хорошо закупоривают баллон, в котором ведется операция). После охлаждения к жидкости прибавляют крахмального раствора и титруют избыток SnCl 2 йодом. Для расчетов необходимо знать титр хлористого олова и отношение между растворами SnCl 2 и йода; отношение это, находимое прибавлением к известному объему йода в присутствии крахмала раствор SnCl 2 до обесцвечивания, и выражается числом куб. см SnCl 2, отвечающих 1 куб. см раствора йода. Что касается титра SnCl 2, то для уменьшения всех могущих быть при этом погрешностей, он устанавливается непосредственно по железу, подобно тому, как для Na 2S2O3. Для этого берут раствор с определенным содержанием Fe 2 Сl 6 и титруют его по вышесказанному, т. е. сначала SnCl 2, затем йодом. Зная сколько пошло йода для обратного титрования SnCl 2 и отношение растворов, можно определить избыток SnCl 2, взятого для реакции и затем легко вычислить, какое количество железа приходится на 1 куб. см SnCl 2 при восстановлении взятой железной соли. Это и будет титр SnCl 2, выраженный непосредственно в железе, и им пользуются для расчетов при анализах. Раствор SnCl 2 готовят, растворяя чистое олово в НCl при нагревании без доступа воздуха и лучше всего в присутствии платины (олово с платиной образует гальваническую пару, и растворение идет энергичнее). Готовят обыкновенно такой раствор, что 1 куб. см его соответствует приблизительно 0,01 железа. Раствор SnCl 2 отличается большим непостоянством и хранится с особенными предосторожностями. Для этой цели лучше всего обычный прибор для титрования соединять с прибором для выделения СО 2, напр. с Кипповским аппаратом, так, чтобы над SnCl 2 постоянно находилась атмосфера этого газа. Надо заметить в заключение, что реакция между SnCl 2 и йодом несколько зависит от концентрации и от большего или меньшего избытка НСl.
Азотная кислота и ее соли. Определение HNO 3 свободной и в солях йодометрическим путем основано на окислении ею в присутствии НCl соединений закиси железа в окисные и сводится на определение этих последних. Реакция между HNO 3 и напр. FeCl 2 идет по уравнению:
2HNO3 + 6FеCl 2 + 6 НCl = 3Fe 2 Сl 6 + 2NO + 4H2O,
и Fе 2Cl6 определяется по первому или второму из вышеописанных способов. Самый процесс раскисления азотной кислоты ведут следующим образом. Готовится раствор FеСl 2, или еще лучше берется двойная железно-аммиачная соль Fe(NH 4)2(SO4)26H2 O (так наз. соль Мора); в продаже она существует в довольно чистом виде и легко сохраняется без изменения: во всяком случае необходимо убедиться в отсутствии в ней окисных соединений железа. Часто бывает достаточно, облив соль водой, дать кристаллам ее раствориться слегка с поверхности, и тогда останется совершенно чистая соль. Для опыта берут небольшой избыток: например около 12 частей на 1 часть KNO 3 ввиду трудности затем удаления NO из подобных растворов. Разложение производят в баллоне от 150 до 200 куб. см емкостью. Поместив в него железную соль, баллон соединяют с прибором для выделения СО 2; вытеснив из баллона воздух, приливают в избытке соляную к-ту уд. веса 1,10—1,12 и бросают навеску HNO 3 или ее соли. Реакция идет при нагревании. Окись азота удаляется из раствора кипячением, причем для ускорения через жидкость пропускают СО 2. По удалении NO, баллон охлаждают, не прекращая тока СО 2, и потом поступают как указано при определении железа.
Белильные соли (eau de Labarraque). Определения в воде жавелевой, Лабаррака и в особенности в белильной извести так наз. активного хлора (хлорометрич. определения) представляют большую важность для завод, техники. Из предложенных для этой цели способов наибольшую простоту представляет способ Р. Вагнера для белильной извести. Он основан на взаимодействии между этой солью и KJ в слабокислом растворе, причем выделяется йод в количестве эквивалентном активному хлору. Если состав белильной извести (см.) выразить формулой СаСl 2 О + Н 2 O, то эта реакция может быть выражена уравнением:
СаС l2 О + Н 2O + 2KJ + 2HCl = CaCl2 + J2 + 2КCl + 2Н 2O.
Опыт ведется следующим образом: берут 10 г хорошо измельченной белильной извести, помещают в толстостенную склянку, обливают водой и, набросав туда кусочков стекла или стеклянных гранат, старательно разбалтывают до тех пор, пока нельзя будет подметить отдельных кусочков соли; после того все это переливают в литровую колбу и разбавляют водой до 1 л (принимая в расчет объем введенного стекла). Для опыта берут 100 куб. см этой жидкости (предварительно хорошо ее взболтав), прибавляют к ней 25 куб. см 10% раствора KJ и при помешивании подкисляют соляной кислотой до слабо кислой реакции; выделившийся йод титруют с помощью Na 2S2O3 обычным способом. Результат выражают или в градусах Гей-Люссака, т. е. сколько литров активного хлора приходится на 1 кг бел. извести, или обычным путем в процентах. Надо заметить, что при этих опытах прибавка большого избытка НCl может повести к ошибкам, так как в белильной извести всегда встречается некоторое количество Са(ClО 3)2, которая с НCl, в свою очередь, может выделить йод из KJ, чего по Вагнеру не случается, если точно следовать его указаниям. Другой способ предложен Пено (Реnоt) и, благодаря Мору, получил широкое применение. Он состоит в окислении хлором (или белильной известью) мышьяковистой кислоты As 2 О 3 в мышьяковую As 2 О 5:
Аs 2O3 + 2 Сl 2 + 2Н 2O = As2O5 + 4HCl;
Аs 2O3 + 2CaCl2O = As2O5 + 2CaCl2.
Применение для этой цели As 2O3 в солянокислом растворе было предложено еще Гей-Люссаком, при чем индикатором служила индиго-серная кислота; этот способ, удержавшийся до сих пор только в одной Франции, состоит в следующем: употребляют раствор, содержащий в 1 литре 4,425 г As 2 О 3 — именно столько, сколько по вышеприведенному уравнению может быть окислено ее в As 2O5 литром хлора, принимая вес последнего при 0° и 760 мм давления равным 3,17344 г. Раствор Аs 2O3 готовится при нагревании ее с соляной кислотой. Для опыта берут пипеткой 10 куб. см этой жидкости, окрашивают ее раствором индиго-серной кислоты в синий цвет и из бюретки приливают сюда до обесцвечивания индиго раствор белильной извести (хорошо приготовленный и заключающий 10 г исследуемой соли в 1 литре). Зная количество израсходованного раствора белильной извести, легко рассчитать, сколько литров активного хлора приходится на 1 кг ее. Неудобство способа заключается в малой чувствительности применяемого здесь индикатора и в том, что в разных условиях для обесцвечивания индиго требуется разное количество хлора. Пено предложил употреблять щелочной раствор As 2 О 3 и индикатором йодокрахмальную бумажку. Берут навеску 4,942 г As 2O3 (по уравнению реакции это количество соответствует 1/10 Сl по весу), приливают около 200 куб. см воды, прибавляют 20—30 г NaHCO 3 или, еще лучше, по Мору, КНСО 3 и кипятят до полного растворения, потом разбавляют до 1 литра. As 2O3 должна быть совершенно чиста и не содержать As 2S3, что иногда можно легко определить, подвергая Аs 2O3 возгонке, если присутствует As 2S3, то будучи летучее As 2O3 он возгоняется в первых порциях, которые являются, таким образом, окрашенными; лучше же, растворив часть As 2O3 в NaOH, пробовать уксуснокислым свинцом, который при As 2S3 дает осадок PbS. Понятно, и в NaHCO 3 или КНСО 3 также не должно содержаться сернистых соединений. Дело в том, что щелочной раствор As 2O3, будучи совершенно постоянным в обыкновенных условиях, в присутствии As 2S3 постепенно окисляется кислородом воздуха, и титр его изменяется. Для опыта берут обыкновенно 10 г белильной извести, растирают ее в ступочке с небольшим количеством воды, смывают потом в литровую колбу и тщательно разболтавши доливают до литра. Для определения берут 50 куб. см этой хорошо перемешанной жидкости, и приливают к ней из бюретки мышьяковистой соли, пробуя время от времени йодокрахмальной бумажкой. Сначала бумажка интенсивно окрашивается выделившимся йодом, а потом, по мере разрушения белильной извести, окраска слабеет и наконец совсем не появляется, что указывает на конец реакции. Определение обыкновенно повторяют с другой порцией раствора белильной извести, причем уже сразу приливают мышьяковистой соли немногим меньше, сколько ее требуется на основании первого опыта и остальное добавляют по каплям, постоянно пробуя йодокрахмальной бумажкой. Самая проба ведется таким образом: погружают стеклянную палочку в исследуемый раствор и потом проводят ею черту на слегка влажной бумаге. Расчет полученных данных не представляет никаких затруднений. Иногда берут навеску не 10 г белильной извести, а 7,1 г, и тогда число куб. см мышьяковистого раствора прямо дает % активного хлора. Способ Пено дает хорошие результаты и удобен потому, что здесь требуется один титрованный раствор, который к тому же прекрасно сохраняется без всяких изменений. Это последнее качество и подало Мору мысль применить мышьяковисто-калиевую соль для всех йодометрических определений вместо Na 2S2O3.
Способ Мора. Основными растворами здесь служат: 1/10 нормальный раствор йода и раствор K 3AsO3, приготовленный как сказано раньше и заключающий 4,932 г As 2O3 в 1 литре. Титр раствора йода определяется по As 2O3. Для этой цели берут некоторое количество раствора K 3AsO3, разбавляют водой, прибавляют крахмального раствора и титруют йодом до появления синего окрашивания. Расчет ведется по реакции
K3AsO3 + 2J + H2O = K3AsO4 + 2HJ.
Конец реакции выступает не особенно резко. Мор нашел, что прибавка небольшого количества углекислого аммония много помогает здесь, и потому он советует постоянно прибегать к этому. Определения по способу Мора, вообще, схожи с тем, что мы имели раньше, при Na 2S2O3; существенной разницей является то, что они ведутся в щелочной среде и если присутствие углекислых солей, щелочных металлов там, как мы видели, ведет к ошибкам, здесь этого нет: K 3AsO3 одинаково относится к свободным галоидам и к их кислородным соединениям типа МХО (где М = К, Na, NH 4, a X = Cl, Br, J), которые, вероятно, образуются здесь в небольшом количестве при титровании. Приведем несколько примеров определения по Мору: а) определение свободного йода ведется в водном растворе KJ и идет настолько чисто, что эта операция может служить для проверки титра K 3AsO3; б) определение хлора в хлорной воде, в белильных солях делается таким образом, что к определенному количеству исследуемой жидкости прибавляют K 3AsO3 в небольшом избытке и потом обратно титруют йодом. При анализе перекисей, СrО 3 и вообще веществ, выделяющих с НCl хлор, последний поглощают раствором Na 2CO3, определяют его как сказано раньше и т. д. В заключение необходимо указать на одно из интереснейших приложений йодометрии — на определение щелочей и кислот. Еще Мор, основываясь на способности кислот выделять J из смеси KJO 3 и KJ, указывал на возможность свести определение кислот (а следовательно и щелочей) к определению йода обычным способом. В этом отношении обстоятельные исследования произвел Грегер (Gr öger, "Zeitschrift für angev. Chemie", 1890 г.). Исходными растворами при анализе у него служат: 1/10 норм. Na 2S2O3 и 1/10 нормальный Н 2 SО 4; кроме того KJ (24 г в 100 куб. см) и KJO 3 (4 г на 100 куб. см). Растворы KJ и KJO 3 должны быть совершенно нейтральны; они хранятся отдельно, и для опыта их берут поровну. Определение титра Na 2S2O3 ведется таким образом, что берут навеску около 0,15 чистой, высушенной при 100° KJO 3; растворив в воде, прибавляют около 0,9 г KJ, приливают потом НCl в небольшом избытке и йод титруют раствором Na 2S2O3. Для установки титра Н 2 SО 4 берут ее известное количество, смешивают с некоторым избытком раствора KJ и KJO 3 и определяют йод серноватисто-натриевой солью; расчет по реакции
5KJ + KJO3 + 3H2SO4 = 3K2SO4 + 6J + 3H2O.
Титрованная Н 2 SО 4 служит для определения щелочей, углекислых солей и пр. При анализе щелочей, напр., к исследуемой жидкости прибавляют Н 2SO4 в небольшом избытке; избыток H 2SO4 определяется по предыдущему, по выделению J из раствора KJ и KJO3. Что касается анализа углекислых солей, то, прибавив избыток титрованной Н 2 SО 4, необходимо, для получения вполне хороших результатов удалить кипячением всю СО 2 из жидкости и уже потом после охлаждения приливать раствор KJ и KJO 3. Дело в том, что СО 2 сама выделяет несколько йод из смеси KJ и KJO 3; во избежание этого и советуют, между прочим, хранить растворы KJ и K JO3. Так же поступают при анализе сернистых металлов. Анализ H 2SO4, HCl, HNO3 ведется таким же образом, как было указано при определении титра Н 2 SО 4; результаты получаются очень хорошие; для органических же кислот способ непригоден, так как здесь выделение йода сильно зависит от концентрации растворов и требует иногда много времени, хотя вероятно, нагревание может ускорить это. Едва ли этот способ представляет преимущество перед обычным определением кислот и щелочей. Гораздо проще решен этот вопрос Бауманом (в "Zeitschrift für angev. Chemie", 1891 г.) и его последователями. Здесь также в основе лежит выделение йода кислотами из смеси KJ в KJO 3; только самое определение йода совершенно другого характера. Опыт показывает, что при действии перекиси водорода Н 2O2 на йод в присутствии щелочи КОН происходит выделение кислорода: именно на 1 молекулу йода 1 молекула кислорода. Реакция идет, вероятно, так:
J2 + 2KOН + H 2O2 = 2KJ + O2 + 3H2O.
Предполагается, что в первый момент действия КОН на йод происходит образование йодноватистой соли:
J2 + 2KOН = KJO + KJ + H 2O,
которая потом с Н 2O2 выделяет кислород
KJO + H2O2 = KJ + H2O + O2,
подобно хлорноватистым и бромноватистым солям. KJO 3 и КСlO 3 этого не показывают. Определив при этой реакции объем выделившегося кислорода, его температуру и давление, мы получаем все данные для расчета. Определение йода по этому способу удобно ведется в приборе, изображенном на фиг. 3. Он состоит из двух частей: реакция между J и Н 2O2 происходит в A, а измерение кислорода в В, А — склянка, емкостью около 150 куб. см, ко дну которой припаян цилиндр около 20 куб. см; при помощи крана и каучука А соединяется с В; В состоит из обыкновенной бюретки в 50 или 100 куб. см, соединяющейся внизу при помощи каучука с манометрической трубкой. Обе трубки на пробках вставлены в стеклянную муфту, наполненную водой, температура которой указывается вставленным в верхнюю пробку термометром; манометрическая трубка имеет внизу отросток и каучуковой трубкой соединяется с сифоном с водой, снабженным каучуковым шаром; M — стакан с водой. Самое определение делается так. В сосуд А наливают 40—50 куб. см исследуемого раствора йода в KJ (если он близок к 1/10 нор. и 60—80 куб. см — если слабее); во внутренний цилиндр наливают около 3 куб. см 2% — 3% раствора Н 2 О 2 и около 5 куб. см раствора КОН (1 ч. на 1 ч. воды) [Опытом найдено, что Н 2 О 2 удобнее брать не более, чем в 2 раза больше того, чем теоретически требуется, а КОН по меньшей мере в 2 раза и не больше, чем в 10 раз. Лучше смешать р-р Н 2 О 2 и КОН в требуемом количестве, охладить до обыкновенной температуры (при смешении выделяется тепло), а потом добавлять в цилиндр].
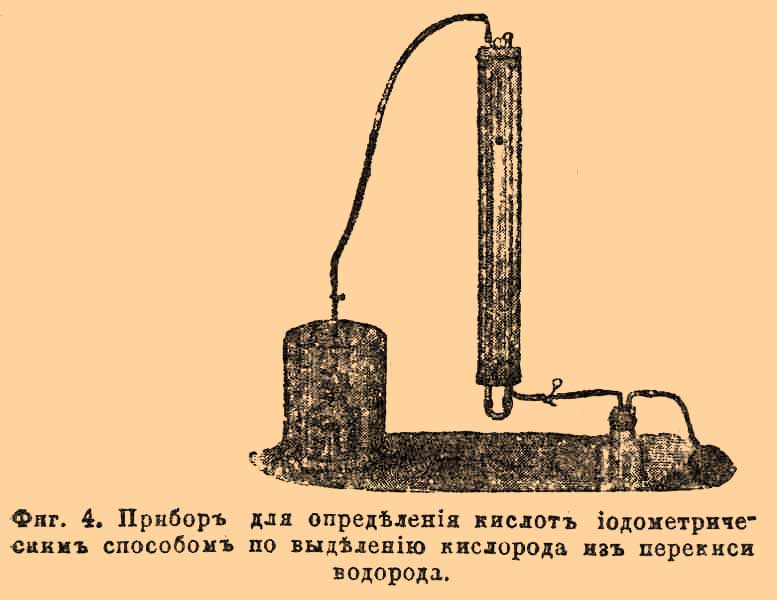
Фиг. 4. Прибор для определения кислот йодометрическим способом по выделении кислорода из перекиси водорода. Потом осторожно вставляют в А пробку и помещают в стакан с водой М. После этого сообщают прибор с атмосферой (вынув кран) и, действуя каучуковым шаром, заставляют воду из сифона подняться в бюретке до 0 и вставляют зажимы. Когда температура в Д уравнялась (температура воды в M и в В приблизительно одинакова), ставят кран на свое место; в приборе таким образом оказывается запертым известный объем воздуха, После этого выливают некоторое количество воды из бюретки, вынимают А из стакана М, осторожно приводят находящуюся в нем жидкость во вращательное движение и потом разом смешивают обе жидкости. Тотчас начинает выделяться кислород. Не переставая вращать, жидкость старательно взбалтывают, потом ставят снова в стакан M и минут через 5—10, приведши воду в бюретке и в манометрической трубке к одному уровню, определяют объем выделившегося кислорода (по разности между вновь наблюденным и прежним, предполагая, что температура и давление не изменялись), замечают и атмосферное давление. По таблицам вычисляют вес кислорода. Опыт кончается довольно быстро. Выделение кислорода продолжается несколько секунд. Ждать с отсчитыванием объема более 1/2 ч. не следует, так как кроме изменения температуры в M и В может произойти выделение кислорода из Н 2 О 2. Для определения кислот Н 2 SО 4, НCl, HNO 3 берут около 2 г KJ и 0,2 KJO3 хорошо измельченного (этого достаточно для получения до 50 куб. см кислорода; если бюретка до 100 куб. см, берут вдвое), помещают в сосуд А, растворяют в небольшом количестве воды и прибавляют известный объем разбавленной кислоты; тотчас выделяется йод; жидкость разбавляют водой до 40—50 куб. см и поступают, как сказано раньше. Результаты вполне удовлетворительны, если придерживаться всех указаний [Первым и необходимым условием для получения хороших данных является полное и быстрое смешивание выделившегося J с Н 2 О 2 и КОН. Дело в том, что KJO, образующ. в первый момент действия КОН на J, крайне непрочна. Она чрезвычайно быстро претерпевает дальнейшее изменение, окисляясь до КJO 3 и не входя во взаимодействие и Н 2 О 2, таким образом результаты получаются ниже, чем следуют.]. Для органических кислот, которые медленно выделяют йод из смеси KJ и KJO 3, реакцию можно ускорить нагреванием; но уже тут теряется та простота, которая отличает этот способ от других.
С. Вуколов. Δ.
|
Page was updated:Saturday, 26-Nov-2016 22:05:56 MSK |