
| [ начало ] | [ К ] |
Крепкая водка*
(хим.-техн.) — старинное, а в настоящее время употребительное в торговле и технике, название азотной кислоты HNO3 (фр. acide nitrique, ас. azotique, нем. Salpeters ä ure, англ. nitric acid). Открытие азотной кислоты относят обыкновенно ко второй половине VIII стол., когда появилось первое описание ее получения с помощью перегонки смеси селитры, медного купороса и квасцов (Гебер). Позднее алхимиками она добывалась разложением селитры при накаливании ее с железным купоросом, мышьяковистой кислотой или глиной и носила различные названия (Aqua dissolutiva, fortis, acuta, acidum nitri и др.). С XVII ст. азотная кислота делается промышленным продуктом, а с середины прошлого ст. Глаубер стал получать ее современным способом, т. е. помощью перегонки селитры с серной кислотой и назвал Spiritus nitri fumans Glauberi. Первые указания на состав азотной кислоты сделаны были Лавуазье (1776), признавшим содержание в ней кислорода, но опытным путем элементарный состав ее из кислорода и азота был доказан впервые лордом Кавендишем (1785), который получил ее синтезом при пропускании электрических искр через воздух в присутствии влаги и щелочей.
В природе азотная кислота в свободном состоянии не встречается, но в соединении с основаниями в виде солей (селитр) распространена, обыкновенно в малых количествах, почти повсюду. В воздухе следы ее содержатся в виде азотноаммиачной соли и образуются частью прямым соединением азота с кислородом в присутствии влаги и аммиака под влиянием электрических разрядов (особенно во время гроз) и разнообразных окислительных процессов, частью окислением самого аммиака (см. ниже). Поэтому она всегда почти находится в дождевой воде и др. атмосферных осадках. В воде озер, рек и источников, попадая в них частью из атмосферы, а главным образом из почвы, находится также в весьма малых дозах, не превышающих нескольких миллиграмм на литр. В несколько больших количествах азотная кислота находится в почвенных водах и в самой почве, где она играет роль первостепенной важности в жизни растений и где образуется, главным образом, на счет окисления кислородом воздуха аммиака, развивающегося при гниении азотистых органических веществ, в присутствии влажности и углекислых солей калия, натрия, магния и кальция, при взаимодействии с которыми и превращается в селитры (см. это сл. и Нитрификация). В некоторых странах (в Остиндии, Туркестане, Перу, Египте и др.) встречаются почвы, весьма богатые селитрами, а в Южной Америке, в смежных частях Чили, Боливии и Перу, именно в бездождной береговой полосе (пустыня Атакама) натровая селитра образует даже богатейшие залежи местами почти чистой соли (см. Селитры). В малых количествах соли азотной кислоты находятся в растениях, а также в моче, поту и др. выделениях животных.
Образование азотной кислоты. Кроме упомянутых выше случаев, азотная кислота образуется окислением азота при взрыве гремучего газа с примесью воздуха, при горении смеси водорода с азотом, а также, в малом количестве, при горении на воздухе водорода, окиси углерода, светильного газа, спирта, стеарина, воска, дерева, угля и др. веществ, при окислении на воздухе фосфора и при электролизе воды, содержащей в растворе воздух. Низшие степени окисления азота [О них см. ст. Окислы азота.], окись азота NO, азотистый ангидрид N 2O3 и двуокись азота NO 2, при достаточном избытке кислорода в присутствии воды, сполна переходит в азотную кислоту. Предварительное образование этих низших степеней окисления доказано также в большинстве приведенных выше случаев синтеза азотной кислоты из элементов. Образование азотной кислоты окислением аммиака, совершающееся, между прочим, как упомянуто выше, и в почве, может происходить при самых разнообразных условиях. Так, оно идет в присутствии щелочей и щелочных земель под влиянием пористых землистых тел, как это показали опыты Дюма и комиссии французских академиков; при пропускании смеси аммиака с кислородом или воздухом через нагретую до 300° трубку с губчатой платиной (весьма энергичная реакция, сопровождающаяся самораскаливанием) или даже просто через сильно раскаленную фарфоровую трубку; при окислении меди воздухом в присутствии аммиака и при действии на аммиак различных окисляющих веществ, как озон, перекиси водорода, марганца, свинца и бария, марганцовокалиевая, двухромокалиевая и бертолетова соли. Во всех этих случаях азотная кислота получается в виде солей, аммиачной или других, и обыкновенно в смеси с солями азотистой кислоты. Из этих солей она легко может быть получена и в свободном состоянии при разложении их кислотами. Так, напр., в водном растворе азотнобариевая соль Ba(NO 3)2 (баритовая селитра, применяемая в пиротехнии и в пороховом деле) с серной кислотой или азотносеребряная соль AgNO 3 (ляпис) с соляной кислотой, разлагаясь по уравнениям: Ba(NO 3)2+H2SO4=2HNO3+BaSO4 и AgNO 3+HCl=HNO3 +AgCl, дают осадки нерастворимых в воде сернобариевой соли и хлористого серебра, а в растворе азотную кислоту.
Получение азотной кислоты в лабораториях и в технике основывается также на разложении солей ее, именно селитр калиевой и натриевой или чилийской, при взаимодействии их с крепкой серной кислотой [Разложение здесь происходит и идет до конца не потому, чтобы серная кислота была более энергичной кислотой, но потому, что она не летуча, а азотная летуча, и по мере своего образования удаляется из круга взаимодействия, благодаря чему вступает в силу закон действия масс (см. Равновесие химическое). Тот же закон действует и при разложении солей азотной кислоты в водных растворах, когда, как в двух приведенных выше случаях, вновь образующаяся при реакции соль выделяется в осадок.]. При умеренном нагревании (до 130°) реакция совершается по уравн., напр. для калиевой селитры: KNO 3+H2SO4 = HNO3 + KHSO4 (1) с образованием кислой сернокалиевой соли рядом с свободной азотной кислотой, которая по своей летучести при этом отгоняется, и останавливается на этой фазе, все равно, будет ли селитра взята в количестве согласно уравн. или в избытке. Если же к концу этой первой фазы температура будет повышена, то при достаточном количестве селитры реакция пойдет далее, по уравн.: KNO 3+ KHSO4 = HNO3 + K2SO4, вследствие чего образуется новое количество свободной азотной кислоты, а в сосуде, где производилось разложение, останется средняя сернокалиевая соль. Таким образом, реакция при высокой температуре совершается по уравн.: 2KNO 3 +Н 2 SО 4=2HNO3+K2SO4 (2). Совершенно то же самое произойдет в обоих случаях, если взять натриевую селитру вместо калиевой, с той лишь разницей, что в остатке будет кислая или средняя сернонатриевая соль. В лабораториях берут большей частью калиевую селитру, с которой масса смеси менее пучится во время реакции и которая обыкновенно имеется в продаже более чистой, и, так как азотная кислота при нагревании, даже незначительно выше температуры ее кипения, уже начинает разлагаться на кислород, воду и двуокись азота, которая, растворяясь в получаемой азотной кислоте, сообщает ей красно-бурую окраску, то, имея в виду непосредственно получение возможно чистого продукта, ведут разложение, согласно первому уравнению, при умеренном нагревании и употребляя на 1 частицу (101 вес. частей) селитры 1 частицу (98 в. ч.) серной кислоты или примерно равные по весу количества обоих веществ. Реакцию производят в стеклянной реторте, а азотную кислоту собирают в охлаждаемой водой или льдом стеклянной колбе-приемнике, по возможности глубже вставляя в нее шейку реторты (рис. см. в ст. Лаборатория).
При добывании азотной кислоты на заводах применяют исключительно чилийскую селитру, которая, примерно, вдвое дешевле калиевой и, сверх того, вследствие меньшого атомного веса натрия (Na 23, K 39), в равном весе содержит больше азотной кислоты, а, следовательно, дает и больший (почти на 20%) ее выход. Относительную пропорцию серной кислоты берут или по уравн. (1), или по ур. (2). Так как получаемая, в качестве побочного продукта, кислая сернонатриевая соль (бисульфат) без предварительной переработки на среднюю соль (см. Сульфат), помимо содовых заводов (см. Сода), сбыта почти не имеет и либо продается за бесценок, либо зачастую и просто выбрасывается, то было бы выгоднее работать согласно ур. (2), расходуя, примерно, вдвое меньшее количество серной кислоты; но, ввиду того, что в этом случае, благодаря высокой температуре реакции, азотная кислота, отчасти разлагаясь, не только получается с большим содержанием низших окислов азота [Это, впрочем, не всегда имеет значение, а иногда даже и желательно, если имеют в виду получение красной дымящей азотной кислоты (см. далее).], которые, кроме того, по своей летучести являются источником значительных потерь в производстве, но и оказывается в общем более слабой [Ибо рядом с крепкой кислотой здесь получается много слабой при уловлении водой низших окислов (см. ниже).], чем при работе по уравн. (1); затем, ввиду того, что получаемый в остатке по уравн. (2) средний сульфат весьма тугоплавок и его, при удалении, приходится выламывать из реторт, затрачивая на это немало времени и труда, тогда как бисульфат, получаемый по уравн. (1), легкоплавок и может быть с большим удобством выпущен в жидком виде — обыкновенно предпочитают добывать азотную кислоту при употреблении избытка серной, особенно, когда стремятся получить ее возможно более свободной от низших окислов азота и в то же время крепкой, как это, напр., требуется для пироксилинового и нитроглицеринового производств. Строго говоря, на практике ни в том, ни в другом случае не придерживаются в точности отношений, требуемых уравнениями (1) и (2), а обыкновенно в одном случае берут на 2NaNO 3 около 1 1/4 Н 2 SО 4 или на 100 в. ч. обыкновенной 96% чилийской селитры 70-75 в. ч. крепкой серной кислоты (купоросного масла) с содержанием 95% моногидрата или в 66° В, а в другом на 2NaNO 3 около 1 3/4 H2SO4 или приблизительно равные количества селитры и серной кислоты. Очень часто для получения слабой азотной кислоты употребляют менее крепкую, а потому и более дешевую серную кислоту в 60-62° В, содержащую от 78 до 82% моногидрата и получаемую сгущением в свинцовых ваннах (см. Купоросное масло), причем на 100 в. ч. чилийской селитры считают от 100 до 110 в. ч. такой кислоты, что приблизительно составляет на 2NaNО 3 около 1 1/2 H2SO4. Представляя преимущество дешевизны, 60 градусная кислота однако сильнее разъедает чугунные сосуды, в которых обыкновенно производится разложение селитры, и требует большего их объема, большего количества топлива и больше времени для перегонки, вследствие чего многие авторитетные заводчики (напр., О. Гутманн в Англии) предпочитают и для приготовления слабой азотной кислоты получать сперва крепкую на крепкой серной кислоте и затем разбавлять ее водой до любой желаемой концентрации. Самая операция разложения селитры прежде производилась в больших стеклянных ретортах, которые размещались в два ряда в соответствующих чугунных или железных котлах на так называемых галерных [Название это произошло от некоторого сходства таких печей с выдающимися по бокам шейками реторт с галерой, спустившей весла на воду.] печах (фиг. 1 и 2).
Фиг. 1. Галерная печь с стеклянными ретортами и сосудами для сгущения азотной кислоты (поперечный разрез).
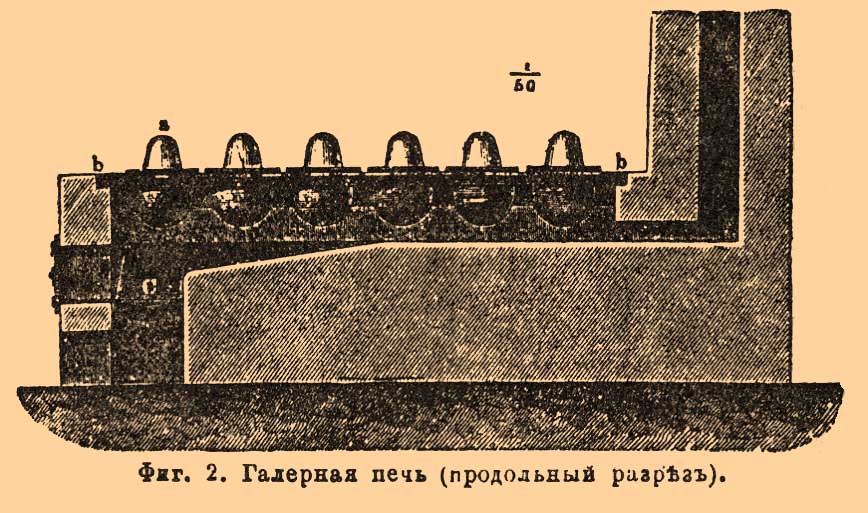
Фиг. 2. Галерная печь (продольный разрез).
По причине ломкости, неудобства заряжания и малой производительности, стеклянные реторты ныне почти вовсе вывелись из употребления и повсюду заменены большими ретортами из чугуна, на который ни крепкая серная кислота, ни пары азотной кислоты почти не действуют. Наибольшим распространением пользуются в настоящее время два типа этих реторт, изображенные в разрезе на фиг. 3 и 4. Старейший, особенно часто употребляемый в Англии тип, представляет лежачие реторты (фиг. 3).

Фиг. 3. Лежачая цилиндрическая реторта.
Они имеют форму открытых с обоих концов чугунных цилиндров A, длиной около 1,5 м, диам. около 0,6 м и с толщиной стенок до 4 см, закрывающихся двумя круглыми массивными чугунными крышками а, прикрываемыми снаружи, с целью предохранения их от потери тепла и от сгущения на них азотной кислоты, круглыми песчаниковыми плитами. Реторты вмазываются в печь обыкновенно попарно и нагреваются топкой С. Трубка d ведет к сосудам ВВ для сгущения азотной кислоты, а свинцовая воронка с служит для введения в реторту серной кислоты. Крышки (иногда с прокладкой из асбестового картона) укрепляются герметически с помощью обыкновенной железн. замазки [100 частей железных опилок, 5 частей серного цвета и 5 частей нашатыря.] или с прибавкой к ней огнеупорной глины и т. п. Крышка, обращенная к сосудам для сгущения, примазывается раз навсегда, другая же, напротив, отнимается для задачи селитры и выгрузки сульфата. При работе с избытком серной кислоты и эту крышку не отнимают, а вводят селитру и выпускают жидкий бисульфат через соответственно устраиваемые в ней отверстия. Количество селитры, задаваемой единовременно в такие реторты, доходит до 305 кг при 240 кг серной кислоты в 66° В, причем гонка длится 16-18 час. Другой тип чугунных реторт, изображенный на фиг. 4, приноровлен исключительно для работы с получением в остатке бисульфата и имеет вид стоячего цилиндрического котла с высотой от 1,2 до 1,5 м и такого же диаметра при толщине стенок до 5 см, способного вмещать от 300 до 600 кг селитры.

Фиг. 4. Стоячая реторта.
Вся реторта находится внутри печной кладки, так что со всех сторон охватывается пламенем, чем достигается меньшая потеря тепла и, след., меньший расход топлива, а главное, это делается для того, чтобы воспрепятствовать сгущению азотной кислоты на верхних частях реторты и тем предохранить их от разъедания. Загрузка реторты селитрой и серной кислотой происходит через верхнюю широкую горловину, герметически запирающуюся с помощью чугунной крышки и цемента из смеси глины с гипсом. Соответствующее отверстие вверху печи плотно прикрывается полой внутри и набитой золой железной крышкой nn. Шейка реторты для защиты чугуна от разъедания сгущающейся азотной кислотой одевается совнутри плотно вмазываемой глиняной трубкой, которая другим своим концом вставляется на замазке в стеклянный форштосс D или иногда сочленяется с холодильником. Для выпуска бисульфата (обыкновенно в соответственно устроенные железные вагонетки) реторта внизу снабжена чугунной трубой, выходящей наружу и на рис. не представленной. Продолжительность гонки при загрузке в 300 кг селитры здесь примерно такова же, как и при лежачих ретортах, а при загрузке в 600 кг доходит до 24-28 часов. При нагревании реторт, содержащаяся в них смесь чилийской селитры с серной кислотой приходит в кипение и при этом столь сильно пенится и вспучивается, что нередко происходит перебрасывание поднимающейся пены через шейку реторты в приемники, особенно если, в стремлении увеличить производительность, чересчур переполняют реторты или сильно их нагревают. Чтобы вполне устранить опасность перебрасывания и в то же время сохранить хорошую производительность, О. Гутманн в Лондоне употребляет реторты очень большого размера, а так как доброкачественная отливка таких реторт целиком была бы весьма затруднительна и стоила бы очень дорого, то он их делает из трех частей (фиг. 5).
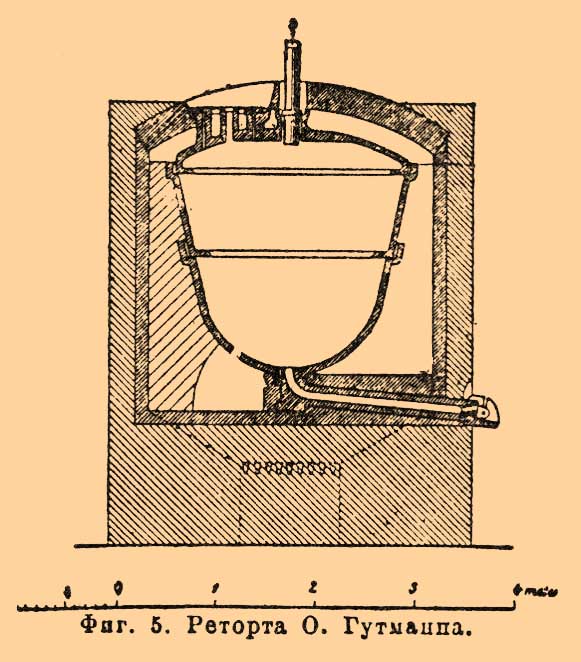
Фиг. 5. Реторта О.Гутманна.
Нижняя, полусферическая часть, сочленяющаяся снизу с чугунной, вмазанной в печную кладку трубой для выпуска бисульфата, служит для вмещения смеси селитры с серной кислотой; средняя кольцеобразная часть назначается исключительно для увеличения внутреннего пространства реторты с целью дать место поднимающейся пене; третья часть составляет крышку с запирающимися отверстиями для введения серной кислоты и селитры и отвода паров азотной кислоты. Крышка [Крышка наиболее подвержена разъедающему действию азотной кислоты и здесь легко и без больших затрат может быть заменена новой, между тем как в ретортах из цельного куска порча верхних частей делает негодной всю реторту.] и средняя часть снабжены выдающимися внутрь реторты фланцами, задерживающими поднятие пены. Все три части скрепляются между собой с помощью огне-и кислотоупорного цемента. В таких ретортах при загрузке селитры в 610 кг О. Гутманн успевает заканчивать перегонку азотной кислоты в течение всего лишь 10-12 час. и, притом, получает кислоту, почти вовсе не содержащую примеси сульфата, серной кислоты и железа (см. ниже). Но такая быстрота перегонки потребовала и устройства специального конденсационного аппарата, так как обычные приемы сгущения (см. ниже) при ретортах Гутманна оказались недостаточными. Обыкновенно для экономии места соединяют вместе по 2 и более печей с ретортами, располагая их в последнем случае то в один ряд, то группами по 4. Остаточным теплом топочных газов пользуются отчасти для предварительного подогревания ближайших к реторте сосудов для сгущения, дабы они не лопались вследствие резкой перемены температуры при вхождении в них первых порций горячей азотной кислоты, ввиду чего в начале перегонки газы из топки, с помощью опускания соответствующей заслонки, направляют по каналу M (фиг. 4), и только лишь, когда сосуды ЕЕ слегка нагреются, заслонку подымают и пускают газы по каналу L; отчасти же для высушивания селитры, что ввиду ее значительной гигроскопичности безусловно необходимо при добывании наиболее крепкой азотной кислоты.
Сгущение паров азотной кислоты чаще всего производится в трехгорлых бутылях (фиг. 4 ЕЕ) или таких же баллонах или бомбонах (bombonnes, tourilles, фиг. 2 е g и фиг. 3 BB) из особой кислотоупорной глины с кранами внизу для выпуска кислоты, соединенных с ретортой большей частью стеклянным форштоссом, а между собой дугообразными глиняными трубами [Соединение производится с помощью эластичной замазки, хорошо противостоящей действию кислот и приготовляемой из тонкого порошка тяжелого шпата на растворе каучука (500 ч .) в льняном масле (2500 ч.) с примесью серы (3 ч.). Другая превосходная, быстро твердеющая в тепле замазка, приготовляется из асбестового порошка в смеси с силикатом натрия.]. Число баллонов изменяется от 7-9 при малых до 16-24 при больших ретортах. Два ряда баллонов от двух реторт обыкновенно замыкаются в конце одной общей глиняной башенкой, наполняемой коксом пли кусками пемзы и орошаемой сверху водой для удержания последних следов азотной кислоты, не успевших сгуститься в баллонах, а главным образом для поглощения NO 2, превращающейся с водой и кислородом воздуха в слабую азотную кислоту, которая и вытекает из башни в подставленный внизу сосуд. Сгущающаяся в баллонах кислота неодинакова по крепости и по чистоте. В первом баллоне она всегда содержит довольно много серной кислоты и сульфата, механически увлекаемых из реторты парами и газами, а также и просто вследствие нередко случающегося перебрасывания содержимого реторты; эту кислоту выливают обыкновенно обратно в реторту. В следующих баллонах получается наиболее чистая и менее всего окрашенная низшими окислами кислота, далее же она содержит хлор, развивающийся на счет примеси к селитре поваренной соли, и в изобилии низшие окислы азота. Иногда при получении слабой азотной кислоты в 36° В, для лучшего сгущения в баллоны наливают немного воды для той кислоты, которая вытекает из башни. Фиг. 6 представляет ныне нередко употребляемый конденсационный аппарат Деверса и Плиссона.
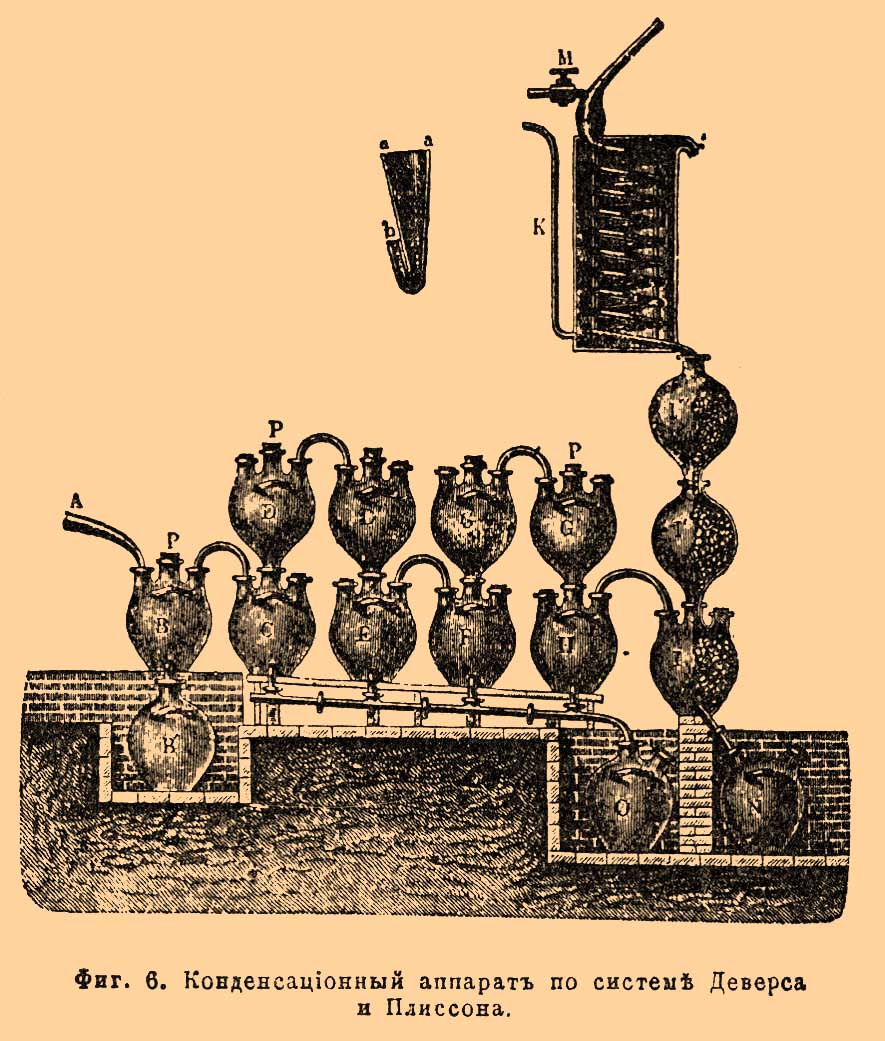
Фиг. 6. Конденсационный аппарат по системе Деверса и Плиссона.
Здесь пары азотной кислоты из реторты поступают в приемник В, сообщающийся с сосудом B', где собирается менее чистая азотная кислота (см. выше). Пары, не сгустившиеся в В, идут, постепенно сжижаясь, последовательно через сосуды С, D, D', E, F, G, G' и H, из которых 4 нижние соединены внизу короткими трубками с общей для всех сосудов наклонной трубой, по которой сгущенная, более или менее чистая азотная кислота течет в приемник О. В наполненных пемзой сосудах J, J', J" и в змеевике К, орошаемых водой через кран М, удерживается остаток паров и NO 2 и в виде слабой азотной кислоты вытекает в приемник N. Иногда воду или слабую кислоту из N понемногу впускают и в сосуды D, D', G, G' через гидравлически запирающиеся воронки Р, представленные на фиг. отдельно при ааb. На заводах серной кислоты нередко NO 2 поглощают крепкой серной кислотой, для чего ставят в конце конденсационного аппарата маленькую Гей-Люссакову башню, и получаемую нитрозу употребляют для питания Гловеровой башни (см. Камерное производство). В настоящее время для более быстрого сгущения азотной кислоты нередко пользуются холодильником в форме змеевика, устраиваемого из глиняной трубки и помещаемого в деревянном баке с проточной водой вслед за первым баллоном (фиг. 7).
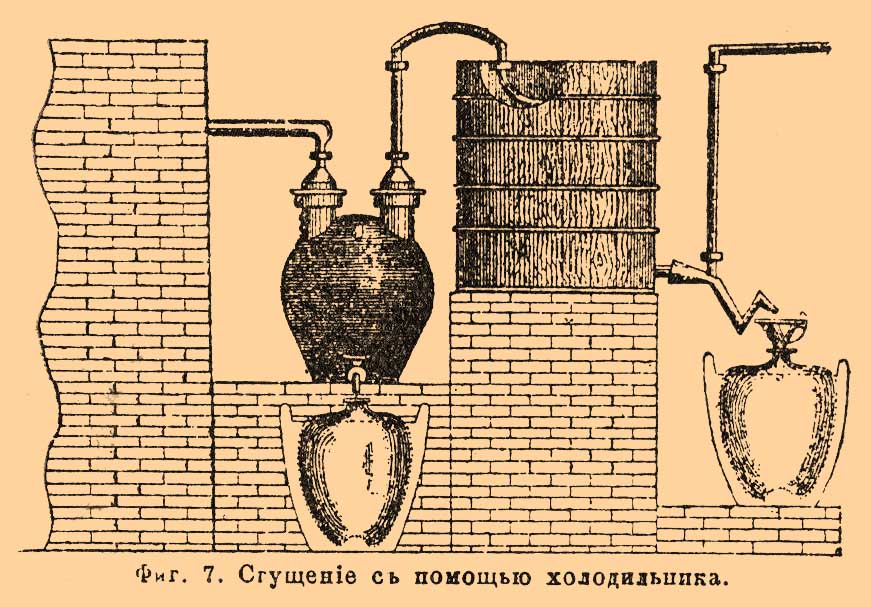
Фиг. 7. Сгущение с помощью холодильника.
Кислота течет из холодильника через коленчатую трубку, препятствующую выходу паров в воздух, прямо в стеклянные бутыли, а остаток паров идет по соответствующей трубке в баллоны и далее в абсорбционную башню. При таком приспособлении, пользуясь тем, что NO 2, сообщающая бурый цвет азотной кислоте, выделяется преимущественно в начале и конце перегонки, представляется возможность собирать почти бесцветную кислоту отдельно от окрашенной. Чаще, впрочем, для получения вполне бесцветной крепкой азотной кисл. [Слабая азотная кислота прямо получается бесцветной вследствие разложения NO 2 водой.], весь перегон подвергают рафинированию или отбелке (белению), для чего его сливают в большой глиняный баллон емкостью до 350 литр. и пропускают через него с помощью насоса струю воздуха при нагревании до 60°. При этой операции, продолжающейся ок. 6 час., воздухом уносится вместе с NO 2, поглощаемой затем в абсорбционной башне, также и вся примесь хлора. В последнее время иногда производят сразу и конденсацию кислоты, и ее отбелку. Так, на химическом заводе в Грисгейме пары азотной кислоты из реторты поступают в двухгорлый баллон, поддерживаемый при температуре в 80°, а из него в восходящий глиняный змеевик, охлаждаемый водой при 30°. Сгущающаяся в змеевике азотная кислота стекает обратно в баллон, а низшие окислы азота через верхний конец змеевика поступают в ряд расположенных вслед за ним баллонов и далее в абсорбционную башню. Пропускание воздуха в баллон, стоящий между ретортой и змеевиком, значительно облегчает выделение NO 2 и позволяет понизить температуру до 60°. Но особенного внимания заслуживает конденсационный аппарат О. Гутманна, изготовляемый на гончарном заводе Л. Рорманна близ Мускау в прусской Силезии.
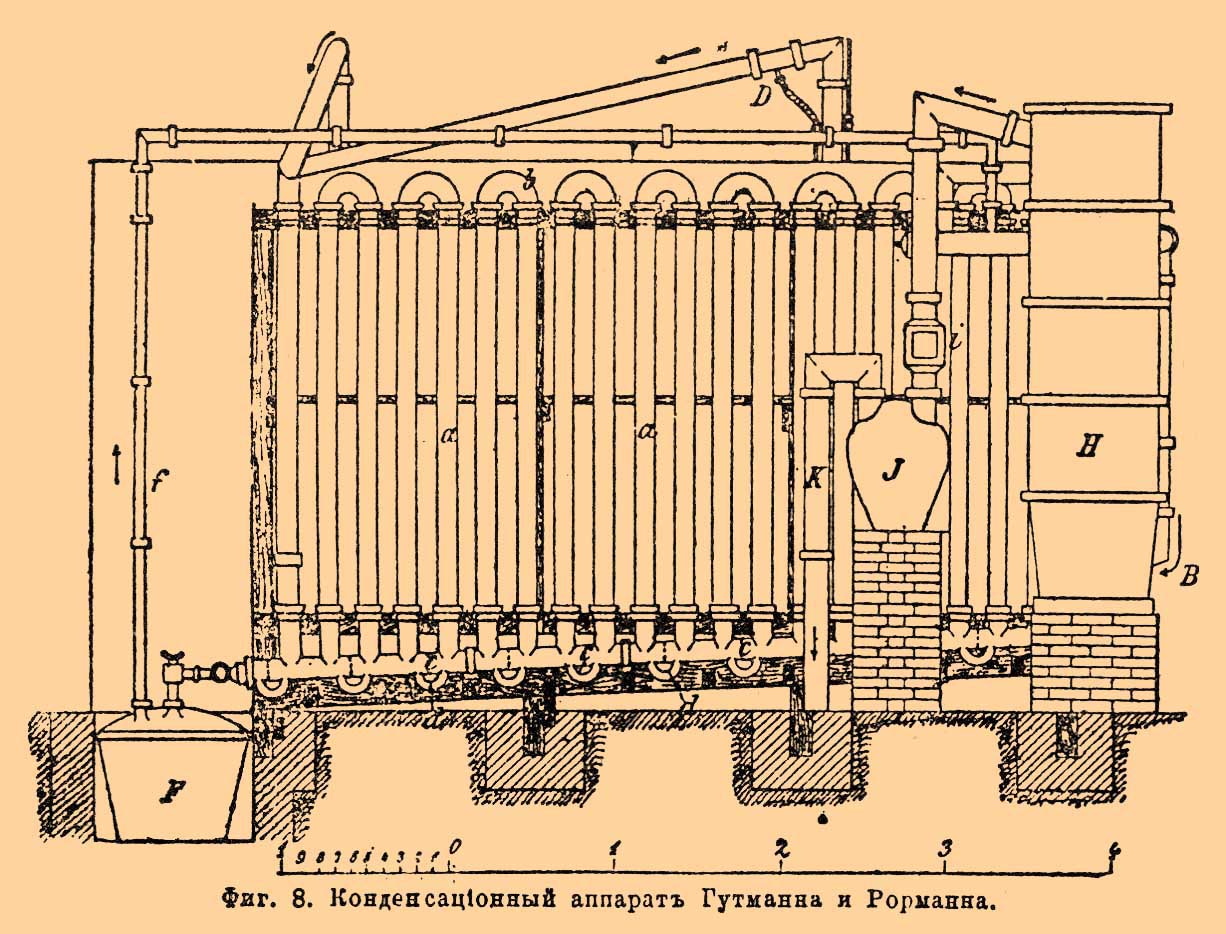
Фиг. 8. Конденсационный аппарат Гутманна и Рорманна.
Как видно из фиг. 8, он состоит для каждой реторты из 20 вертикальных глиняных труб ааа..., длиной в 2,5 м и с толщиной стенок всего в 8 мм, соединенных вверху попарно дугообразными глиняными же трубками, а внизу сообщающихся между собой с помощью несколько наклонной трубы, разделенной на короткие камеры ссс... поперечными перегородками, на фиг. обозначенными пунктиром, таким образом, что пары и газы не могут проникать из одной камеры в другую и двигаться непосредственно по трубе ссс..., а непременно должны проходить зигзагообразно по вертикальным трубам ааa... Камеры ссс... сообщаются между собой лишь маленькими дугообразными трубками ddd..., по которым сгущающаяся в ааа... и стекающая вниз азотная кислота переливается непрерывно из камеры в камеру, образуя в то же время собой гидравлический запор между камерами, и течет в приемник F, служащий одновременно для двух аппаратов, располагаемых параллельно [На фигуре виден лишь один, ближайший к зрителю.]. Реторты, находящиеся в печи A, сообщаются каждая с соответствующим аппаратом посредством глиняных труб, в которые, с помощью инжектора D, вдувается подогретый до 80° воздух, служащий частью для непосредственного превращения в самом аппарате низших окислов азота с присутствующими парами воды в азотную кислоту, частью же для выдувания их вместе с хлором из сгущающейся в аппарате кислоты и вытеснения в орошаемую водой абсорбционную башенку H и далее в баллон J, где они и удерживаются в виде слабой азотной кислоты. Главнейшие выгоды Гутманн-Рормановского аппарата (в связи с указанным выше усовершенствованным типом реторт) заключаются в том, что он, с одной стороны, благодаря большой охлаждающей поверхности и потому быстроте сгущения, допускает вдвое более быструю гонку, чем обыкновенно, и, с другой стороны, дает азотную кислоту с очень малым содержанием NO 2 (редко более 1%), вовсе не содержащую хлора, более крепкую (95-96% моногидрата) и в почти теоретическом выходе. Кроме того, он весьма мало занимает места и количество слабой кислоты (40°В.), получающейся в абсорбционной башне, составляет при нем лишь от 3 до 7% всего выхода (считая на HNO 3), тогда как с обычными аппаратами оно даже в лучших случаях редко менее 10%, при общем выходе в 94% теоретического (см. ниже). В самое последнее время (1893) Гутманн и Рорманн сократили число труб ааа... до 5 (вместо 20) и окружили их холодильником в виде деревянного ящика с проточной водой, через что количество слабой кислоты уменьшилось до 2%, но зато крепость главной массы кислоты понизилась до содержания 94-95% моногидрата и несколько увеличилось содержание в ней NO 2. В той или другой форме конденсационный аппарат Гутманна и Рорманна пригоден также при денитрации отработавших кислотных смесей с пироксилиновых и динамитных заводов и, по уверению авторов, особенно практичен при добывании азотной кислоты разложением селитры с помощью этих смесей и вообще более слабой серной кислоты. Об устройстве абсорбционной башни H (Plattenthurm, патент Лунге-Рорманна), составляющей необходимую часть всего аппарата, см. Соляная кислота.
Собирающуюся в приемниках азотную кислоту разливают в толстостенные (непременно) стеклянные бутыли (флаконы) с пришлифованными стеклянными пробками, вместимостью около двух пудов, в которых она и поступает в продажу. Бутыли обертываются соломой и упаковываются в плетеные из ивовых прутьев корзины. Так как в случае разбития бутыли разлившаяся азотная кислота, даже и не крепкая (36°В), особенно в теплое и сухое время, легко может произвести воспламенение упаковки, то последнюю нередко пропитывают раствором какой-либо соли, напр. глауберовой, серно-магнезиальной и т. п.
Выход азотной кислоты. Теоретически, согласно уравнению (см. выше), 85 кг NaNO 3 должны дать 63 кг HNO 3 или 100 кг NaNO 3 74,118 кг HNO 3. Так как продажная чилийская селитра содержит обыкновенно от 94 до 98% чистой соли и от 2 до 6% посторонних примесей (хлористого натрия, сернонатриевой соли, воды и землистых веществ), то теоретический выход из нее будет несколько меньшим, а именно 100 кг дадут от 69,7 (при 94%) до 72,6 (при 98%) кг HNO 3 или в среднем (при 96%) 71,2 кг HNO 3, что составит 134,8 кг азотной кислоты в 36°В. (с 52,8% HNO 3). В действительности выход в таком размере никогда не достигается вследствие того, что малые количества азотной кислоты частью удерживаются сульфатом в реторте, а частью уходят в дымовую трубу, в виде низших окислов азота, не успевших поглотиться водой в абсорбционной башне. Эти потери (по данным Лунге, Сореля и др.), при употреблении обычных аппаратов, в общем составляют обыкновенно от 4 до 8%, так что выход моногидрата HNO 3 обыкновенно колеблется между 92 и 96% теоретического. Таким образом, при хорошей работе, считая потерю в 6%, 100 кг NaNO 3 (96%) дадут 66,9 кг HNO 3 или 126,7 кг кислоты в 36°В. При добывании концентрированной кислоты с содержанием HNO 3 в 90% и выше слабая азотная кислота, получаемая в абсорбционной башне в количестве не менее 10% общего выхода, также, можно сказать, составляет потерю, которая в данном случае достигает в сумме 16% и более (относительно результатов работы с аппаратом Гутманна-Рорманна см. выше). Что касается расхода угля, то он обыкновенно принимается в 1/2 пд. на каждый пуд селитры.
Продажная азотная кислота и ее очищение. Получаемая описанным выше [Из других способов добывания азотной кислоты укажем лишь на некоторые и, между прочим, на способ, предложенный Кульманом (1863) и основанный на разложении селитры при накаливании ее (230°) с хлористым марганцем по уравн. 5MnCl 2 + 10NaNO3 = 2Mn2O3+MnO2+10NaCl+10NO2+O2. При пропускании газообразных продуктов реакции с прибавкой воздуха в конденсационную башню с водой, NO 2 дает азотную кислоту крепостью в 35°В и в почти таком же выходе, как и при разложении селитры с помощью серной кислоты. Способ преимущественно применим на заводах, добывающих хлорную известь (см.), где может отчасти служить для так называемого оживления закиси марганца с той выгодой, что вместо выбрасываемого хлористого кальция здесь будет получаться поваренная соль, дающая сульфат и соляную кислоту, и, след., хлор будет полнее утилизироваться, а известь и вовсе не будет расходоваться. Подобным же образом селитра разлагается и при накаливании ее с хлористыми или сернокислыми цинком, магнием и даже кальцием. Вагнер, для получения азотной кислоты, предложил накаливание селитры с кремнеземом или гидратом глинозема: 2NaNO 3+3SiO2 = Na2Si3O7+2NO2 +О и 6NaNO 3+Al2(OH)6 = Al2(ONa)6 +6NaNO3, причем в первом случае в качестве побочного продукта получается растворимое стекло (см.), а во втором алюминат натрия, дающий при разложении углекислотой соду и снова глинозем. Фогт и Вихман (1893), накаливая смесь селитры с известью, мелом или окисью железа или марганца в струе угольной кислоты и водяного пара, получают в конденсационном аппарате азотную кислоту и в виде побочного продукта соду.] путем и обращающаяся в торговле азотная кислота представляет собственно различной крепости водные растворы моногидрата, отвечающего формуле HNO 3, причем преимущественно на заводах готовят эти растворы трех концентраций, именно в 86°, 42-43° и 48° Б. Первый, который собственно и носит название крепкой водки (Scheidewasser, Acidum nitricum), бесцветен, имеет уд. в. около 1,33, содержит около 53% HNO 3 и готовится либо разбавлением водой более крепкой кислоты, либо перегонкой селитры с 60° В. серной кислотой, причем в приемники наливается немного воды. Азотная кислота в 42-43° В. или двойная крепкая водка, тоже бесцветна, уд. в. около 1,42, содержит около 70% HNO 3 и, след., близка по составу к постоянно кипящему гидрату (см. ниже). Она непосредственно получается при перегонке селитры с 60-62 градусной серной кислотой. Кислота в 48° Б. представляет дымящую азотную кислоту (Acidum nitricum fumans) с содержанием до 94% HNO 3 и с уд. в. около 1,50. Столь крепкая азотная кислота, хотя и может быть получена вполне бесцветной с помощью отбелки, но редко бывает такой, ибо легко разлагается в прикосновении с частицами случайно попадающих в нее органических веществ (пыли), от нагревания и даже от действия света с образованием NO 2, которая, растворяясь, и окрашивает ее в цвета от желтого до более или менее темно-оранжевого. Количество NO 2 в ней большей частью не превышает, однако, 3-4%. Для получения ее селитру высушивают и берут купоросное масло в 65-66° В. и обыкновенно в избытке. Кроме этих сортов, в продаже имеется еще так наз. красная дымящая азотная кислота, представляющая обыкновенную дымящую кислоту, но с большим содержанием NO 2 в растворе. Она обыкновенно получается при перегонке в лежачих ретортах 2 мол. селитры с 1 мол. крепкой серной кислоты, когда значительная часть азотной кислоты разлагается по уравнению: 2HNO 3 = 2NO2+H2 O+O. Иногда для содействия такому разложению — в реторту, на каждые 100 частей селитры, прибавляют 3 1/2 части крахмала, который и производит раскисление азотной кислоты. Последняя в этом случае оказывается очень богатой низшими окислами азота, содержит кроме NO 2 еще и N 2O3, имеет темно-бурый или (от примеси N 2O3) зеленовато-бурый цвет и при своем получении требует хорошего охлаждения приемников. Обыкн. красная кислота, в зависимости от содержания в ней HNO 3 и количества NO 2, имеет уд. вес от 1,50 до 1,55. Продажная крепкая азотная кислота, кроме низших степеней окисления азота, содержит нередко весьма малую примесь железа, серной кислоты и сульфата, механически увлекаемых из реторт во время перегонки, и почти всегда следы хлора, а иногда и йода. От низших окислов ее очищают на заводах, как было указано выше, с помощью процесса отбелки, причем удаляется также и хлор; для освобождения от других примесей азотную кислоту иногда подвергают вторичной перегонке с прибавкой небольшого количества чистой селитры для того, чтобы связать свободную серную кислоту; примеси при этом остаются в перегонном аппарате. Йод частью удаляется вместе с хлором, частью же остается при перегонке вместе с другими примесями в виде йодноватой кислоты. В лабораториях иногда освобождают азотную кислоту от низших окислов, переводя и их в азотную кислоту окислением двухромовокалиевой солью, которая при этом переходит в соль окиси хрома, а затем перегоняя при возможно низкой температуре, лучше всего в пустоте. Для получения безводной азотной кислоты, отвечающей составу гидрата HNO 3 [Собственно кислоты, точно отвечающей такому составу, еще никому не удалось получить, а наиболее безводная содержит 98,8% HNO 3 и 0,2% воды (Roscoe).], чистую и возможно более крепкую азотную кислоту осторожно перегоняют в стеклянной реторте на водяной бане с равным или двойным объемом крепкой серной кислоты, которая и удерживает воду, а также частью и NO 2 [По уравнению: 2NO 2+H2SO4 = (HSO З)(NO)O+HNO3 ], причем собирают лишь первые порции перегона, переходящие при температуре 86°.
Состав и свойства азотной кислоты. Чистый гидрат (нормальный или мета-гидрат) азотной кислоты HNO 3 (см. выше примечание) содержит 1,59% водорода, 22,22% азота и 76,19% кислорода, имеет частичный вес 63 и представляет чрезвычайно едкую, бесцветную жидкость уд. в. при 15°/4° = 1,5204 (Lunge 1891, для кислоты с 99,7% HNO 3) и при 0° = 1,559 (Kolb 1886, для кислоты с 99,8% HNO 3), застывающую при-47° и кипящую при 86°. Безводная, равно как и К. азотная кислота, содержащая менее 25% воды, дымит на воздухе вследствие того, что легко летучий и испаряющийся уже при обыкнов. темп. гидрат HNO 3, соединяясь с влажностью воздуха, образует гидрат менее летучий (см. ниже), с меньшей, чем у воды, упругостью пара, а потому и сгущающийся в форме видимого глазу тумана (дыма). В отсутствии воды и в крепких растворах HNO 3 является веществом столь мало прочным, что разлагается не только от нагревания, но даже и от действия света с выделением кислорода и NO 2 (см. выше). Теоретическая плотность паров азотной кислоты, отвечающая формуле HNO 3, по отношению к воздуху = 2,18; опытом найдены (Carius 1871) следующие плотности, при t 86°-2,05, при t 100°-2,02, при t 130°-1,92; а при t 256° наступает уже полное разложение паров азотной кислоты по уравн.: 2HNO 3 = 2NO2 + H2 O + O и плотность пара тогда = 1,25 (теор. 1,20). Из этих данных следует, что уже при темп. кипения около 9,5% паров азотной кислоты оказываются разложенными на кислород, воду и двуокись азота. Присутствие избытка водяных паров препятствует такому разложению, вследствие чего разбавленная водой азотная кислота перегоняется, не разлагаясь. Главнейшие термохимические данные об азотной кислоте, отнесенные к граммовой ее частице и к жидкому состоянию, сведены в прилагаемой таблице:
|
Томсен. |
Бертело. |
|
| Теплота образования из элементов (H,N,O 3) |
+41,51 |
+41,6 кал |
| Теплота образования из ангидрида и воды 1/2(N2O5H2O) |
— |
+ 7,1 кал. |
| Теплота образования из двуокиси азота 1/2(N2O4, O, H2O) |
+9,335 |
— |
| Теплота образования из окиси азота 1/2(2NO, O3, H2O) |
+ 28,905 |
— |
| Теплота растворения (HNO 3, aq) |
+7,58 |
+7,15 кал. |
| Скрытая теплота плавления |
— |
0,6 кал. |
| Скрытая теплота испарения |
— |
7,2 кал. |
Азотная кислота смешивается с водой во всех пропорциях с значительным, как видно из таблицы, отделением тепла. Все растворы азотной кислоты в воде имеют уд. в. меньший и кипят при более высокой темп., чем безводная кислота (ср. Серная кислота), а более разбавленные, кипят даже при высшей темп., чем вода. Наивысшую темп. кипения имеет раствор уд. в. 1,405-1,424, содержащий около 70% HNO 3 и кипящий при нормальн. атмосф. давлении при 121°-123°. Если перегонять слабую азотную кислоту, то в приемник сперва переходит, главным образом, вода и темп. кип. постепенно повышается, пока крепость кислоты в перегонном аппарате не достигнет 68%. В это время темп. в парах доходит до 121° и остается без изменения во все остальное время перегонки, причем дестиллат получает одинаковый состав с перегоняемой кислотой. Тот же самый результат, т. е. кислота с 68% HNO 3 и с постоянной темп. кип. 121°, получается и при перегонке К. кислоты. В этом случае также происходит постепенное повышение темп. кип., но вначале гонится почти безводная кислота. Постоянство, хотя и не вполне строгое, темп. кип. и большое понижение упругости пара заставляют видеть в рассматриваемом растворе определенное химическое соединение HNO 3 с водой. Дальтон, Бино, Смит выражают его состав формулой 2HNO 3.3H2 O, требующей содержания 70% HNO 3 и отвечающей составу некоторых солей азотной кислоты, напр. Cu(NO 3)3CuO. Д. И. Менделеев, на основании изменения свойств производной ds/dp [ds есть приращение уд. в . в зависимости от изменения % состава на dp.], принимает существование гидрата HNO 3.2H2O = N(HO)5, содержащего 63, 64% HNO 3 и застывающего при-19°, и считает, как и Вислиценус, постоянную температуру кип. 121° за темп. разложения этого гидрата. Бертело, на основании тепловых явлений, наблюденных им при разбавлений водой азотной кислоты различной концентрации (но оспариваемых, впрочем, Томсеном), также признает гидрат HNO 3.2H2 O. Собственно, постоянно кипящий гидрат азотной кислоты не удовлетворяется ни той, ни другой формулой, так как, по Роско, он содержит 68% HNO 3. Кроме того, Роско показал, что состав его меняется в зависимости от давления, при котором производится перегонка, а также и от температуры. Так, при давлении в 70 мм он содержит 66,6%, при 150 мм 67,6%, при 735 мм 68% и при 1220 мм 68,6% HNO 3, а при испарении кислоты с помощью продувания сухого воздуха получается, независимо от состава исходной кислоты, при 13° кислота с 64%, при 60° с 64,5% и при 100° с 66,2% HNO 3. Кроме HNO 3.2H2 O, Д. И. Менделеев, основываясь на изменении уд. веса, указывает на необходимость признания по крайней мере еще одного гидрата, именно HNO 3.5H2 O, отвечающего содержанию 41,2% HNO 3. Приводим (в сокращенном виде) таблицу уд. весов растворов азотной кислоты с указанием также крепости их по ареометрам Боме и Twaddel'я, данную Лунге и Реем (1891 [Точность определений, положенных в основание этой таблицы, дается авторами такая: для состава ±0,02%, для уд. в. ± 0,0001]), числа которых большей частью близко совпадают с числами Кольба (1866), отклоняясь лишь для крепких растворов.
|
Уд. вес при 15°/4° испр. на взвеш. в возд. |
Градусы по Боме. |
Градусы Twaddel'я |
100 вес. ч. coдepжaт | |
|
N2 О 5 |
HNO3 |
|||
|
1,320 |
35,0 |
64 |
43,47 |
50,71 |
|
1,330 |
35,8 |
66 |
44,89 |
52,37 |
|
1,3325 |
36,0 |
66,5 |
45,26 |
52,80 |
|
1,340 |
36,6 |
68 |
46,35 |
54,07 |
|
1,350 |
37,4 |
70 |
47,82 |
55,79 |
|
1,360 |
38,2 |
72 |
49,35 |
57,57 |
|
1,370 |
39,0 |
74 |
50,91 |
59,39 |
|
1,380 |
39,8 |
76 |
52,52 |
61,27 |
|
1,3833 |
40,0 |
— |
53,08 |
61,92 |
|
1,390 |
40,5 |
78 |
54,20 |
63,23 |
|
1,400 |
41,2 |
80 |
55,97 |
65,30 |
|
1,410 |
42,0 |
82 |
57,86 |
67,50 |
|
1,420 |
42,7 |
84 |
59,83 |
69,80 |
|
1,430 |
43,4 |
86 |
61,86 |
72,17 |
|
1,440 |
44,1 |
88 |
64,01 |
74,68 |
|
1,450 |
44,8 |
90 |
66,24 |
77,28 |
|
1,460 |
45,4 |
92 |
68,56 |
79,98 |
|
1,470 |
46,1 |
94 |
71,06 |
82,90 |
|
1,480 |
46,8 |
96 |
73,76 |
86,05 |
|
1,490 |
47,4 |
98 |
76,80 |
89,60 |
|
1,495 |
47,8 |
99 |
78,52 |
91,60 |
|
1,500 |
48,1 |
100 |
80,65 |
94,09 |
|
1,505 |
48,4 |
101 |
82,63 |
96,39 |
|
1,510 |
48,7 |
102 |
84,09 |
98,10 |
|
1,515 |
49,0 |
103 |
84,92 |
99,07 |
|
1,520 |
49,4 |
104 |
85,44 |
99,67 |
Азотная кислота окрашивает лакмус сперва в яркий кирпично-красный цвет и затем обесцвечивает; она представляет одну из самых энергичных минеральных кислот. По количеству тепла 13,7 кал., отделяемого граммовым ее эквивалентом при нейтрализации таким же эквивалентом сильной щелочи (едкого натра) в разведенных растворах, она одинакова с галоидоводородными (за исключением HF) кислотами, уступая в этом отношении лишь серной, селеновой, ортофосфорной и фтористоводородной кислотам, по жадности же ( = 1) занимает вместе с соляной кислотой первое место. Будучи одноосновной кислотой, она образует лишь один ряд солей, состав которых выражается общей формулой M(NO 3)n. Кислых солей в общепринятом смысле для нее неизвестно, но основные довольно многочисленны. Азотнокислые соли обыкновенно получаются при действии азотной кислоты на металлы (см. ниже), их окислы или углекислые соли; они могут образоваться также в водных растворах при взаимодействии азотной кислоты и с другими солями или при двойных разложениях азотнокислых солей с солями других кислот. Последний способ, напр., применяется в обширных размерах в технике для получения обыкновенной калиевой селитры из чилийской и хлористого калия: KCl+NaNO 3 = KNO3 +NaCl (так называемая конверсионная селитра), а также для получения азотно-аммиачной соли из калиевой или баритовой селитры и серноаммиачной соли. Характерная особенность солей азотной кислоты состоит в том, что все они растворимы в воде и большей частью легко. Из основных солей, напротив, большинство в воде трудно растворимо; такова, напр., применяемая в медицине основная азотновисмутовая соль Bi(OH) 2NO3 (Magisterium bismuthi). Все соли азотной кислоты мало прочны при высокой температуре и потому при нагревании более или менее легко разлагаются, подобно самой азотной кислоте, с выделением свободного кислорода. Характер разложения при этом зависит как от температуры, так и от природы основания, заключающегося в соли. Так, соли щелочных металлов при нагревании несколько выше температуры плавления выделяют лишь 1/3 кислорода, переходя в соли азотистой кислоты; при дальнейшем накаливании выделяется новое количество кислорода и свободный азот, а в остатке получается окись металла. Соли щелочноземельных и тяжелых металлов выделяют при накаливании низшие окислы азота и кислорода оставляя окиси (напр. Са (NO 3)2, Pb (NO3)2), перекиси (Mn(NO 3)2) или металл (AgNO 3). Легкость выделения кислорода обусловливает окислительное действие азотнокислых солей при высокой температуре на многие тела. Уголь, сера и горючие органические вещества в смеси с азотнокислыми солями чрезвычайно энергично сгорают при накаливании или в прикосновении с огнем, давая в известных условиях вспышку или взрыв. Отсюда применение азотнокислых солей (по преимуществу KNO 3) в пороховом деле (см. Порох). Подробности о солях азотной кислоты см. при соответствующих металлах, а также в ст. Ляпис, Селитры. Подобно другим кислотам, азотной кислоте свойственно при взаимодействии со спиртами и иными веществами спиртового характера, заключающими в своем составе водный остаток ОН, образовать сложные эфиры (см.) по общему уравн.: R(OH) n + nHNO3 = R(NO3)n + nH2 O. Таковы, напр., азотнометиловый СН 3(NO3) и азотноэтиловый С 2H5(NO3) эфиры, получаемые при действии азотной кислоты на древесный и винный спирты в присутствии азотнокислой мочевины, азотноглицериновый эфир или так наз. нитроглицерин С 3H5(NO3)3 (см.), нитроклетчатка или пироксилин (см.) и др. Последние получаются при действии на холоде дымящей азотной кислоты на глицерин, клетчатку и пр. в присутствии избытка концентрированной серной кислоты, служащей для поглощения выделяемой при реакции воды (см. уравн.). Эфиры азотной кислоты представляют большей частью энергичные взрывчатые вещества (см.). При действии азотной кислоты или смесей ее с серной на углеводороды и многие их производные она их нитрует (см. Нитрование), образуя особый ряд веществ, так наз. нитросоединения (см.). Особенно хорошо известны и легко образуются нитросоединения тел ароматического ряда. Таковы нитроуглеводороды, напр., нитробeнзoл C 6H5(NO2), двyнитpoбeнзoл C 6H4(NO2)2, нитронафталин С 10 Н 7 (NO2), нитрофенолы, напр. тринитрофенол или пикриновая кислота C 6H2(NO3)3 HO и др. Нитросоединения, по крайней мере высшие продукты нитрации, подобно азотным эфирам, суть также взрывчатые вещества, но отличаются по своему химическому строению, ибо в то время как в азотных эфирах остаток азотной кислоты NO 2 или нитрогруппа замещает водород водной группы НО, в нитросоединениях та же нитрогруппа оказывается замещающей атомы водорода углеводородного остатка, как это ясно видно на примере пикриновой кислоты.
Большое содержание в азотной кислоте кислорода (более 76%) и легкость, с которой происходит его выделение (см. выше), обусловливают чрезвычайно энергичную окисляющую способность азотной кислоты по отношению ко многим веществам, вследствие чего она является одним из важнейших и наиболее часто применяемых в практике окислителей. Сера, селен, йод, фосфор, мышьяк окисляются азотной кислотой в кислоты серную, селенистую, йодноватую, фосфорную и мышьяковую. Окисление фосфора крепкой азотной кислотой идет столь энергично, что сопровождается его воспламенением. Уголь, предварительно накаленный, горит в парах азотной кислоты, как в чистом кислороде. На водород при обыкн. темп. азотная кислота не действует, но в присутствии нагретой губчатой платины или при накаливании, напр. при пропускании его вместе с парами азотной кислоты через накаленную трубку, а также в момент его выделения из других соединений, окисляет его, образуя воду. Галоидоводородные кислоты окисляются азотной кислотой с выделением свободных галоидов I, Br и Cl. Если влить небольшое количество слегка подогретой дымящей азотной кислоты в сосуд, наполненный газообразным йодистым водородом, то реакция происходит чрезвычайно эффектно, сопровождаясь появлением большого пламени и отделением фиолетовых паров йода. Сернистый водород превращается крепкой азотной кислотой в серную кислоту, а сернистые металлы — в сернокислые соли. Низшие степени окисления металлоидов и металлов переводятся азотной кислотой в высшие. Так, сернистая кислота, фосфористая и мышьяковистая переводятся в кислоты серную, фосфорную и мышьяковую, а закиси железа и олова — в соответствующие окиси. Из металлов лишь золото, платина, родий, иридий, тантал и титан не изменяются азотной кислотой, все же прочие в тех или иных условиях ею окисляются. Если образующиеся при этом металлические окислы имеют характер оснований, то они, при дальнейшем взаимодействии с азотной кислотой, превращаются в азотнокислые соли, и явление окисления сопровождается растворением металла в азотной кислоте. Так, напр., при действии азотной кислоты на медь образуется азотномедная соль по уравн.: 3Cu+8HNO 3 = 3Cu(NO3)2 + 2NO +4H2 O и жидкость окрашивается в присущий этой соли голубой цвет. Олово, сурьма, молибден, вольфрам азотной кислотой не растворяются, но превращаются ею в белые, аморфные осадки мета-оловянной, сурьмяной, молибденовой и вольфрамовой кислот [Слабая азотная кислота в отсутствии нагревания, впрочем, растворяет олово, ибо в этом случае образуется растворимая в воде, весьма непрочная азотнокислая соль закиси олова Sn(NO 3)2.]. Обыкновенно, чем крепче азотная кислота, тем энергичнее ее действие на металлы, однако не во всех случаях. Так, концентрированная азотная кислота на железо, свинец и серебро, а чистый гидрат HNO 3 при об. темп. также и на медь, олово и висмут почти не действуют, между тем как разбавленная водой растворяет их с большой легкостью. При железе это зависит от того, что оно, под влиянием К. азотной кислоты, приобретает так наз. пассивное состояние (см. Железо), при свинце же и серебре обусловливается нерастворимостью в К. азотной кислоте азотнокислых солей этих металлов, которые, раз образовавшись с поверхности металла и оставаясь на ней в виде тонкого, плотного слоя, предохраняют металл от дальнейшего действия на него кислоты.
Окислительное действие азотной кислоты на органические вещества отличается чрезвычайным разнообразием в зависимости от их природы, концентрации кислоты и температуры. Разбавленная водой азотная кислота действует вообще более или менее умеренно, в большинстве случаев не разрушая частиц окисляемых тел. Напр., винный спирт переводит в альдегид, уксусную, гликолевую, щавелевую кислоты и др. продукты, глицерин в глицериновую, сахар в сахарную, углеводород толуол в бензойную кислоту, обесцвечивает синее индиго, превращая его в изатин и т. п. Концентрированная азотная кислота, за исключением тех условий, при которых она нитрует или превращает в азотные эфиры (см. выше), на большинство органических тел, особенно при нагревании, производит более глубокое окислительное действие, сопровождающееся более или менее полным разрушением их частиц и превращением их большей частью в воду, угольную и щавелевую кислоты. При этом реакция сопровождается столь большим отделением тепла, что нередко происходит воспламенение, как, напр., при действии дымящей азотной кислоты на скипидар, солому, шерсть или др. легко горючие вещества. Сюда же относятся и случаи воспламенения и взрывов при нитровании хлопка и глицерина на пироксилиновых и динамитных заводах. При нагревании в запаянных трубках, след., под давлением, азотная кислота вполне разрушает все органические вещества, окисляя их в воду и углекислоту, между прочим и те, которые содержат серу и галоиды, что и применяется при количественном определении последних в органических веществах (способ Кариуса). Кожу, шерсть, рог и др. азотистые органические тела азотная кислота окрашивает сперва в желтый цвет и затем вполне разрушает. На живом теле она также производит желтые пятна и трудно заживающие ожоги и раны.
Количество кислорода, которое отдает азотная кислота при всех этих реакциях окисления, зависит от ее концентрации, температуры, природы окисляемого тела и др. условий. В большинстве случаев 2 частицы HNO 3 отдают 3 атома О, раскисляясь сами до окиси азота NO по уравн.: 2HNO 3=H2O+2NO+O3; но часто раскисление азотной кислоты может ограничиваться образованием из нее двуокиси азота NO 2 или азотистого ангидрида N 2O3 [Образование этих соединений, а также и NO, которая с кислородом воздуха дает NО 2, обусловливает появление удушливых бурых паров при большей части реакций окисления, производимых азотной кислотой.], или же, наоборот, идти далее до закиси N 2 O и свободного азота N и даже сопровождаться восстановлением в аммиак NH 3 и гидроксиламин NH 3 O. Так, напр., при окислении йода и бромистого водорода образуется NO 2, при окислении йодистого водорода NO, при окислении фосфора NO и N. Сернистый газ SO 2 раскисляет крепкую HNO 3, а также в присутствии крепкой серной кислоты до N 2O3; при избытке SO 2 и возвышенной температуре раскисление идет до NO, а при избытке воды или слабой серной кислоты до N 2 O (ср. Камерное производство). Соли закиси железа превращают HNO 3 в NO, хлористое олово в NH 3 O и NH 3. При окислении металлов, смотря по металлу и условиям реакции, образуются NO 2, N2O3, NO, N2 O и N. Монтемартини (1892) приводит в связь характер раскисления при этом азотной кислоты со способностью металлов разлагать воду с выделением водорода. Действительно, его исследования, а также данные, ранее известные, позволяют вообще принять, что металлы, не выделяющие водорода из воды, напр., такие, как серебро, медь, ртуть, висмут и др., раскисляют азотную кислоту преимущественно лишь до NO 2, N2O3 и NO, тогда как цинк, кадмий, железо, олово, отчасти свинец, т. е. вообще все способные разлагать воду с выделением водорода, подвергают азотную кислоту более глубокому раскислению, превращая ее по преимуществу в NO, N 2 O и N, а также и восстановляют ее далее в NH 3, а олово и в NH 3 O. Со всей строгостью такого разделения металлов, однако, провести нельзя. Что касается щелочных и щелочноземельных металлов, то они при действии HNO 3 частью выделяют свободный водород, частью образуют NH 3 (Bloxam 1869; Montemartini). Достойно внимания наблюдение Велея (Vеlеу 1891), что 30% азотная кислота, вполне свободная от содержания азотистой, при обыкн. темп. не действует на медь, ртуть и висмут, но в присутствии даже весьма малых количеств азотистой кислоты растворение этих металлов происходит легко [По прежним наблюдениям Миллона (1843), подобным же образом относится к разведенной азотной кислоте серебро, а также и многие другие металлы.]. Вообще содержание низших степеней окисления азота NO 2 и N 2O3 в азотной кислоте значительно повышает окислительную способность последней. Поэтому красная дымящая азотная кислота является вообще более энергично действующим окислительным средством, чем чистая азотная кислота. Но в некоторых случаях, благодаря тому, что NO 2 и N 2O3 способны сами окисляться, переходя в HNO 3, она, наоборот, действует восстановительно, отнимая кислород от богатых им веществ, напр. от хромовой и марганцовой кислот, которые переводит при этом в соли окиси хрома и закиси марганца.
Применения азотной кислоты. Она является необходимым элементом трех крупнейших отраслей современной химической промышленности, именно: производства серной кислоты (см. Камерное производство), взрывчатых веществ и искусственных органических красок. Камерное производство потребляет главную массу азотной кислоты, именно около 30% всей добычи ее на земном шаре, включая в это число и ту ее часть, которая добывается непосредственно в каналах серных и колчеданных печей (см. Камерное производство). Применение в технике взрывчатых веществ обнимает собой производство различных видов нитроклетчатки [Из них коллодионная применяется также в фотографии, медицине и для выделки целлюлоида (см.).], нитроглицерина, гремучей ртути, пикриновой кислоты и нек. др. нитропроизводных ароматического ряда. В производстве искусств. органич. красок азотная кислота служит для получения нитробензола [Под названием мирбановой эссенции нитробензол применяется также в парфюмерии.], из которого затем готовится так наз. анилиновое масло, нитротолуола и пр., азотнометилового эфира, употребляемого ныне вместо дорогостоящего йодистого метила при метилировании розанилинов, и мышьяковой кислоты (из мышьяковистой), применяемой для окисления анилинового масла. Кроме того, она непосредственно применяется в красильном деле: для окраски в желтый цвет кожи, шерсти, шелка, рога и др. азот содержащих веществ; в ситцепечатании — для вытравления желтого рисунка по синему фону ткани, окрашенной индиго; для приготовления железной протравы при окрашивании шелка в черный цвет; для получения желти Марциуса и ализарин-оранжа и пр. Далее азотная кислота идет для производства азотнокислых солей: азотносеребряной или ляписа (в медицине и фотографии), азотновисмутовой (мед.) и др.; для вытравления рисунков на меди и стали в граверном деле; для окрашивания золота; для обработки латуни и бронзы (бронзирование); для отделения серебра от золота; для очищения ртути; для приготовления царской водки (см.); для растворения ртути при амальгамировании цинков, для гальванических элементов и для мн. др. разнообразных применений, являясь в том числе одним из важнейших реагентов в химической лабораторной практике. Мировое производство азотной кислоты превосходит ныне 100000 тонн в год и в последнее время сильно возрастает, отчасти благодаря открытию и введению для вооружения армий бездымного пороха. Так, в 1880 г. оно составляло 49850 тонн, а в 1890 г. достигла уже 98595 тонн, из которых около 3/4 приходится на долю Европы и 1/4 — на долю Североамериканских Соединенных Штатов [В эти числа не вошло количество азотной кислоты, добытое в России; но оно вообще не велико и существенно изменить их не может.].
Анализ азотной кислоты. Для распознавания азотной кислоты свободной или в виде солей [В последнем случае к испытуемому раствору прибавляется серная кислота для выделения азотной в свободном состоянии.] в растворах можно пользоваться действием ее на металлы, напр. медь, причем выделяются бурые пары низших окислов азота, или обесцвечиванием слабого раствора синего индиго при нагревании (см. выше), но гораздо более чувствительны следующие реакции. 1) Раскисление железным купоросом в NO по уравн.: 2KNO 3+6FeSO4 + 4H2SO4 = 2NO + 3Fe2(SO4)3+K2SO4+4H2 O и образование соединения его с NO темно-бурого цвета (см. Железо). Испытуемый раствор смешивается в пробирке с крепкой серной кислотой и, по охлаждении смеси, к ней приливается осторожно, чтобы жидкости не смешались, раствор FeSO 4; тогда на границе разделения слоев жидкостей появляется бурое окрашивание, исчезающее при нагревании или взбалтывании пробирки. 2) Выделение йода из йодистого кадия. Азотная кислота сама но себе йода из йодистого калия не выделяет (отличие от азотистой кислоты), но выделяет его в присутствии цинка, вследствие восстановления ее при этом в азотистую. Реакция ведется на холоде в присутствия крахмального клейстера, дающего с йодом интенсивное синее окрашивание и позволяющего открыть 0,001% азотной кислоты в растворе. 3) Синее окрашивание с раствором дифениламина в крепкой серной кислоте представляет самую чувствительную реакцию на азотную кислоту. При производстве опыта одну или несколько капель испытуемого раствора прибавляют к раствору дифениламина в крепкой серной кислоте. Кроме того, в качестве чрезвычайно чувствительных реакций применяются: красное окрашивание с бруцином в присутствии крепкой серной кислоты и желтое с фенолсерной кислотой в присутствии аммиака (проба Шпренгеля). Для распознавания азотной кислоты в твердых солях ее может быть применено выделение некоторыми из них бурых паров низших окислов азота при накаливании в запаянной с одного конца стеклянной трубке. В присутствии окиси свинца окислы азота выделяются при накаливании всеми солями HNO 3. Вспышка с углем или другими горючими телами также может служить для характеристики азотной кислоты. В отличие от солей хлорноватой кислоты, дающих подобную же реакцию, соли азотной кислоты превращаются при этом в углекислые соли, окислы или металлы, соли же хлорноватой кислоты дают хлористые металлы. Так как большинство описанных реакций свойственно также и азотистой кислоте, то они являются доказательными лишь при доказанном отсутствии последней (см. Окислы азота).
Количественное определение. Содержание свободной азотной кислоты в растворах легко может быть найдено по удельному весу, пользуясь приведенной выше таблицей. Столь же легко оно определяется в отсутствии других кислот объемным путем с помощью титрования едким натром (ацидиметрически, см. Анализ объемный). Для определения весовым путем нейтрализуют свободную азотную кислоту аммиаком, раствор выпаривают и по высушивании при 100° взвешивают полученную азотноаммиачную соль NH 4NO3. Способы определения азотной кислоты в ее солях весьма разнообразны. Определение из потери основано на разложении солей азотной кислоты кремнекислотой при прокаливании их с чистым кварцем. Опр. титрованием щелочью. Азотнокислую соль подвергают перегонке (лучше в пустоте) с умеренно концентрированной серной кислотой, перегоняющуюся азотную кислоту собирают в приемнике с отмеренным количеством титрованного раствора едкого натра, где затем количество ее узнают обратным титрованием щелочи серной кислотой. Азотнокислые соли оснований, сполна осаждаемых щелочами, осаждают избытком титрованного раствора NaHO, применяя и здесь метод обратного титрования [Общие основания подобных и других, упоминаемых далее объемных определений, способы расчета числовых данных и практические подробности см. в ст. Анализ объемный, Оксидиметрия.]. На способности азотной кислоты окислять соли закиси железа в соли окиси, по уравн.: 6FeCl 2+6HCl+2HNO3 = 3Fe2Cl6 + 2NО + 4H 2 O, основано несколько способов ее определения в азотнокислых солях. В одних из этих способов количество ее узнается (с помощью уравнения реакции) по количеству окислившейся соли закиси, в других — по количеству образовавшейся окиси азота NO. В способе, открытом Пелузом и разработанном Фрезениусом, берется точно определенное количество соли закиси железа, по произведении окисления оставшийся неокисленным избыток ее определяется титрованием хамелеоном и количество окисленной соли узнается из разности. В способе Брауна непосредственно определяется количество образовавшейся соли окиси железа титрованием хлористым оловом или в комбинации его с йодом (см. Йодометрия). При определении HNO 3 по количеству NO (способ Шлезинга и многочисленные его видоизменения) последнюю собирают над ртутью или крепким раствором едкого натра и либо, переводя ее затем с помощью кислорода или перекиси водорода в азотную кислоту (2NO + О 3 + Н 2O = 2HNO3), титруют едким натром, либо непосредственно измеряют в виде газа по объему в цилиндре, разделенном на куб. см. При всех этих способах, для избежания окисления соли закиси железа или NO кислородом воздуха, реакция ведется в отсутствии последнего, для чего его вытесняют из приборов водяным паром, углекислотой или водородом. При определении по объему NO, воздух вытесняют водяным паром или углекислотой и последнюю потом поглощают едким кали. Весьма точный и удобный способ определения по объему выделяющейся NO представляет раскисление азотнокислых солей ртутью в присутствии крепкой серной кислоты в нитрометре (см.). Наконец, имеется целый ряд способов, основанных на восстановлении азотной кислоты в аммиак NH3 (эквивалент NH 3 соответствует эквиваленту HNO 3). Восстановление производится в колбочке водородом в момент его выделения при взаимодействии смеси цинковых и железных опилок со щелочью (раствор едкого кали уд. в. 1,3) и затем дело сводится к определению аммиака, производимому чаще всего титрованием, для чего аммиак отгоняется кипячением щелочного раствора в приемник, содержащий отмеренное количество титрованной серной или соляной кислоты, избыток которой обратно титруется щелочью. Можно вести восстановление и в кислом растворе, лучше всего оловом с 20% соляной кислотой, полученную аммиачную соль разлагать затем щелочью и поступать далее по предыдущему. Для определения азотнокислых солей в воде часто применяют удобный, хотя и не вполне точный, способ титрования раствором индиго в присутствии серной кислоты.
Испытание продажной азотной кислоты. Присутствие хлора узнается известным образом с помощью азотнокислого серебра (см. Соляная кислота), присутствие серной кислоты (см.) с помощью хлористого бария. Йод, который кипячением испытуемого образца азотной кислоты (с целью удаления низших окислов азота) переводится в йодноватую кислоту, открывается с помощью чистого йодистого калия, который сам не должен содержать йодноватой кислоты, и крахмала на основании реакции: HJO 3 + 5KJ + 5HNO3 = 5KNO3 + 3J2 + 3H2 O (см. Йод). Присутствие низших окислов азота видно по цвету азотной кислоты. Количественно они определяются проще всего титрованием хамелеоном (см. Окислы азота).
Азотный ангидрид N2O5 = 2HNO3 — H2 O. Выше было показано, что, подвергая крепкую азотную кислоту перегонке с крепкой серной кислотой, можно отнять всю воду азотной кислоты, за исключением той, которая входит в состав ее гидрата HNO 3. Эта же последняя удерживается в гидрате HNO 3 столь прочно, а связь азота с кислородом в нем столь слаба, что почти во всех случаях распадение его с выделением кислорода и образованием низших окислов азота наступает ранее разложения его на воду и отвечающий ему ангидрид N 2O5. Поэтому долгое время полагали, что азотный ангидрид вовсе неспособен к самостоятельному существованию, пока в 1849 г. С. Клер-Девиллю не удалось получить его с помощью разложения азотносеребряной соли хлором при нагревании (50°-60°) по уравн.: 2AgNO 3 + Cl2 = N2O5 + 2AgCl + О. Позднее Вебер дал способ получения азотного ангидрида и непосредственно из гидрата HNO 3, отнимая от него воду осторожным действием фосфорного ангидрида (2HNO 3 + P2O5 = N2O5 + 2НРО 3) на холоде и отгоняя затем образовавшийся азотный ангидрид при умеренном подогревании. Перегон, собирающийся при этом в охлаждаемом водой приемнике и содержащий, кроме азотного ангидрида, жидкий гидрат состава N 2O5.2HNO3 или 2N 2O5.H2 O (двуазотная кислота [Этот гидрат получен также Вебером соединением азотн. ангидрида с азотн. кислотой. Он жидок при обыкновенной температуре, застывает при 5°, имеет уд. в. 1,642 (при 18°), дымит на воздухе и легко разлагается со взрывом.]) и низшие окислы азота, представляет бурую жидкость, состоящую из двух несмешивающихся между собой слоев, из которых верхний, темнее окрашенный, при повторном вымораживании выделяет вполне чистый азотный ангидрид в кристаллическом виде. Если при этом, по Бертело, взять фосфорного ангидрида по весу лишь немного более, чем азотной кислоты, и вести как самую реакцию, так и отгонку при возможно низкой температуре, то азотный ангидрид получается в хорошо охлаждаемом приемнике непосредственно в форме больших белых кристаллов, и только под конец перегонки в приемник переходит некоторое количество упомянутой выше двуазотной кислоты. Азотный ангидрид представляет высшую степень окисления азота [Готфейлем и Шапюи, при действии тихого разряда на смесь азота с кислородом, и Бертело, при действии индукционного тока на смесь двуокиси азота и кислорода, получен очень непрочный и еще более богатый кислородом окисел азота — надазотная кислота, в виде жидкости, обладающей характером перекисей. Состав ее с точностью не установлен, но, вероятно, отвечает формуле NO 3, или, по Менделееву, N 2O7.]. Он кристаллизуется в блестящих и прозрачных ромбических призмах уд. в. около 1,64, плавящихся при 30° и перегоняющихся, отчасти разлагаясь, при 45°-50°. При хранении азотный ангидрид постепенно разлагается, быстрее на прямом солнечном свету, а при нагревании иногда со взрывом, на 2NO 2 +O, с водой жадно соединяется, превращаясь в азотную кислоту, расплывается на воздухе и чрезвычайно энергично окисляет органические и мн. др. тела, но на большинство металлов, напр. на олово, магний, свинец, таллий, медь, железо, не действует. Теплота его образования из элементов в газообразном состоянии отрицательная и равна-0,6 кал. (Бертело). О низших степенях окисления азота — двуокиси азота NO 2, азотистом ангидриде N 2O3 и отвечающей ему азотистой кислоте HNO 2, окиси азота NO, закиси азота N 2 O и азотноватистой кислоте HNO — см. Окислы азота.
П. П. Рубцов. Δ.
|
Page was updated:Tuesday, 11-Sep-2012 18:15:40 MSK |